Recine Federica, Bongiovanni Alberto, Casadei Roberto, Pieri Federica, Riva Nada, De Vita Alessandro, Mercatali Laura, Liverani Chiara, Spadazzi Chiara, Miserocchi Giacomo, Fausti Valentina, Amadori Dino, Ibrahim Toni
Osteoncology and Rare Tumors Center, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) IRCCS, Meldola Department of Orthopedics, Istituto Ortopedico Rizzoli, University of Bologna, Bologna Pathology Unit, Morgagni-Pierantoni Hospital, Forlì, Italy.
Medicine (Baltimore). 2017 Nov;96(45):e8545. doi: 10.1097/MD.0000000000008545.
Leiomyosarcoma (LMS) is a malignant sarcoma that can occur in different anatomic sites, including the bone, showing similar histological characteristics but heterogeneous clinical behavior and prognosis. Primary bone LMS was first described in 1965. It is a very rare sarcoma, accounting for <0.7% of all primary malignant bone tumors.
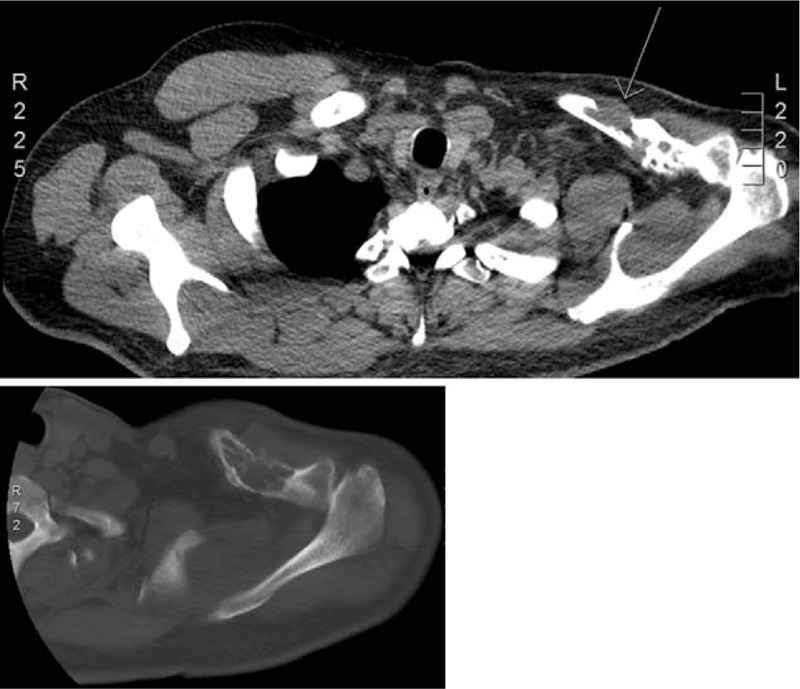
We report the case of a 52-year-old male with primary bone LMS who presented with a solitary osteolytic lesion with focal cortical destruction in the left clavicle, seen on an x-ray and subsequent computed tomography (CT) scan.
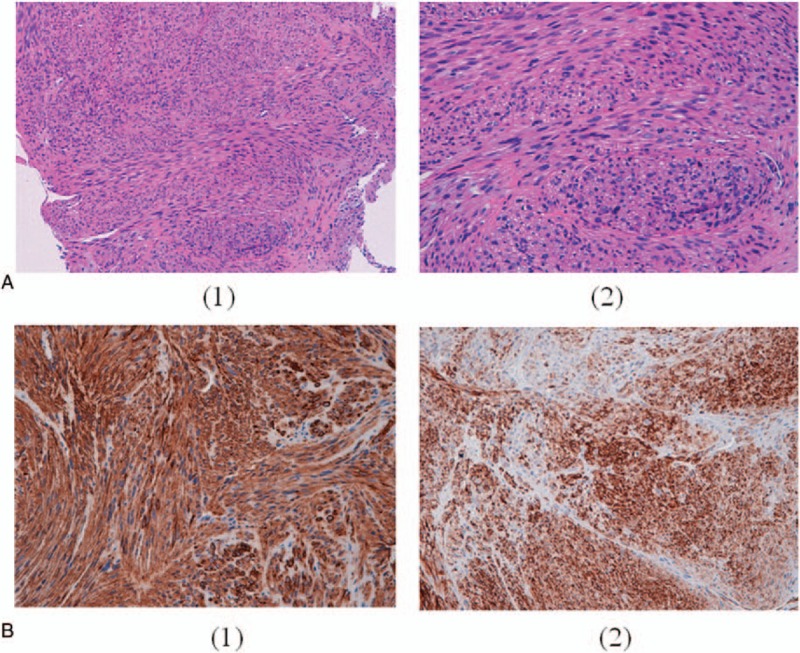
The multidisciplinary Osteoncology team of our institute planned a biopsy that revealed the presence of spindle and pleomorphic cells with a positive reaction for smooth muscle actin and desmin at immunohistochemical analysis, without the presence of cartilage or bone matrix. These results were consistent with a high-grade malignant LMS arising from the bone.
Complete surgical resection of the tumor was performed and a decision was made with the patient not to proceed with adjuvant chemotherapy or radiotherapy.
After more than 1 year of surgery, the patient is well, with no evidence of recurrent or metastatic disease. Follow-up is ongoing.
Little is known about the biology and clinical behavior of bone LMS due to its extreme rarity. A multidisciplinary team in a specialized center is needed for the optimal management of the disease. Surgery with a curative intent is the cornerstone of treatment of localized disease. No data are available about chemotherapy in neoadjuvant, adjuvant, or advanced settings. Further research is needed to identify more effective therapies.
平滑肌肉瘤(LMS)是一种恶性肉瘤,可发生于不同解剖部位,包括骨骼,其组织学特征相似,但临床行为和预后存在异质性。原发性骨平滑肌肉瘤于1965年首次被描述。它是一种非常罕见的肉瘤,占所有原发性恶性骨肿瘤的比例不到0.7%。
我们报告了一例52岁男性原发性骨平滑肌肉瘤患者,X线及后续计算机断层扫描(CT)显示其左锁骨有一个孤立的溶骨性病变,伴有局灶性皮质破坏。
我们研究所的多学科骨肿瘤团队计划进行活检,免疫组织化学分析显示存在梭形和多形性细胞,平滑肌肌动蛋白和结蛋白呈阳性反应,且不存在软骨或骨基质。这些结果与起源于骨的高级别恶性平滑肌肉瘤一致。
对肿瘤进行了完整的手术切除,并与患者决定不进行辅助化疗或放疗。
手术后1年多,患者情况良好,无复发或转移疾病迹象。随访正在进行中。
由于骨平滑肌肉瘤极为罕见,对其生物学特性和临床行为了解甚少。对于该疾病的最佳管理需要专业中心的多学科团队。以治愈为目的的手术是局限性疾病治疗的基石。关于新辅助、辅助或晚期情况下化疗的数据尚无可用。需要进一步研究以确定更有效的治疗方法。