Laboratory of Anthropology, Genetics and Peopling History, Department of Genetics and Evolution - Anthropology Unit, University of Geneva, Geneva, Switzerland.
Institute of Genetics and Genomics in Geneva, University of Geneva, Geneva, Switzerland.
HLA. 2018 Jan;91(1):36-51. doi: 10.1111/tan.13180.
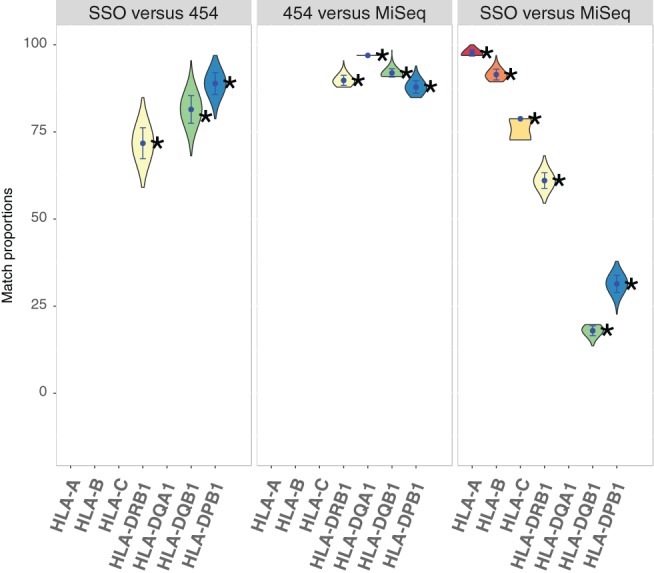
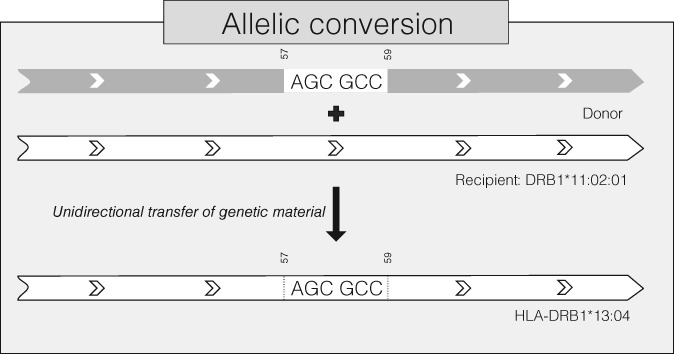
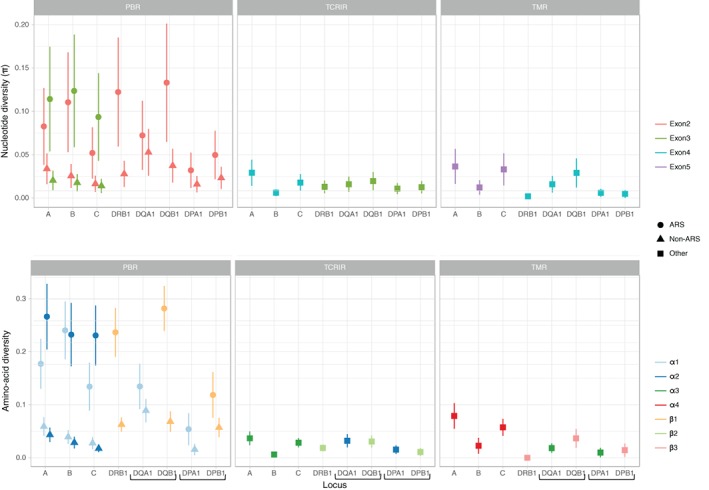
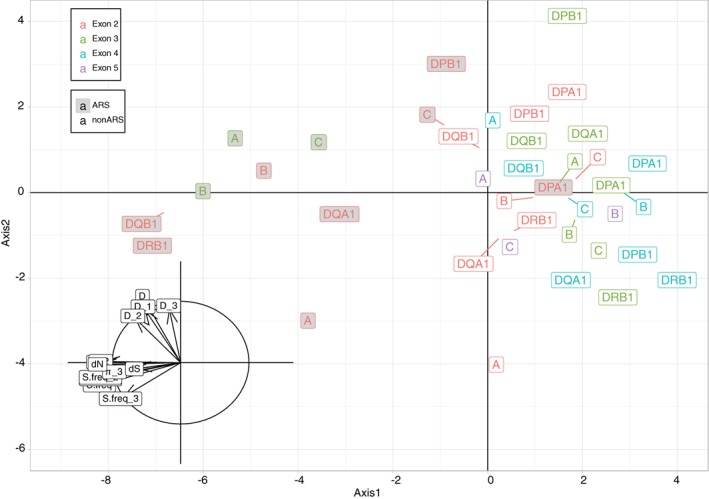
With the aim to understand how next-generation sequencing (NGS) improves both our assessment of genetic variation within populations and our knowledge on HLA molecular evolution, we sequenced and analysed 8 HLA loci in a well-documented population from sub-Saharan Africa (Mandenka). The results of full-gene NGS-MiSeq sequencing compared with those obtained by traditional typing techniques or limited sequencing strategies showed that segregating sites located outside exon 2 are crucial to describe not only class I but also class II population diversity. A comprehensive analysis of exons 2, 3, 4 and 5 nucleotide diversity at the 8 HLA loci revealed remarkable differences among these gene regions, notably a greater variation concentrated in the antigen recognition sites of class I exons 3 and some class II exons 2, likely associated with their peptide-presentation function, a lower diversity of HLA-C exon 3, possibly related to its role as a KIR ligand, and a peculiar molecular diversity of HLA-A exon 2, revealing demographic signals. Based on full-length HLA sequences, we also propose that the most frequent DRB1 allele in the studied population, DRB113:04, emerged from an allelic conversion involving 3 potential alleles as donors and DRB111:02:01 as recipient. Finally, our analysis revealed a high occurrence of the DRB113:04-DQA105:05:01-DQB1*03:19 haplotype, possibly resulting from a selective sweep due to protection to Onchorcerca volvulus, a prevalent pathogen in West Africa. This study unveils highly relevant information on the molecular evolution of HLA genes in relation to their immune function, calling for similar analyses in other populations living in contrasting environments.
为了了解下一代测序(NGS)如何提高我们对人群中遗传变异的评估和对 HLA 分子进化的认识,我们对来自撒哈拉以南非洲(曼登卡)的一个有充分记录的人群中的 8 个 HLA 基因座进行了测序和分析。全基因 NGS-MiSeq 测序的结果与传统分型技术或有限测序策略的结果进行比较,结果表明,位于外显子 2 之外的分离位点对于描述不仅是 I 类,而且是 II 类人群多样性至关重要。对 8 个 HLA 基因座的外显子 2、3、4 和 5 的核苷酸多样性进行全面分析,揭示了这些基因区域之间的显著差异,特别是在 I 类外显子 3 和一些 II 类外显子 2 的抗原识别位点集中的更大变异,可能与其肽呈递功能有关,HLA-C 外显子 3 的多样性较低,可能与其作为 KIR 配体的作用有关,以及 HLA-A 外显子 2 的独特分子多样性,揭示了人口统计学信号。基于全长 HLA 序列,我们还提出,在所研究的人群中最常见的 DRB1 等位基因 DRB113:04 是由涉及 3 个潜在供体等位基因和 DRB111:02:01 作为受体的等位基因转换产生的。最后,我们的分析揭示了高发生率的 DRB113:04-DQA105:05:01-DQB1*03:19 单体型,可能是由于对在西非流行的寄生虫 Onchorcerca volvulus 的保护作用而产生的选择压力。这项研究揭示了与 HLA 基因免疫功能相关的分子进化的高度相关信息,呼吁在生活在不同环境的其他人群中进行类似的分析。