Department of Biology, University of Virginia, Gilmer Hall 044, Charlottesville, VA, 22904, USA.
Center for Quantitative Sciences, Vanderbilt University, Nashville, TN, 37232-6848, USA.
BMC Genomics. 2017 Nov 22;18(1):898. doi: 10.1186/s12864-017-4306-1.
Cowpea (Vigna unguiculata (L.) Walp.) is the most important food and forage legume in the semi-arid tropics of sub-Saharan Africa where approximately 80% of worldwide production takes place primarily on low-input, subsistence farm sites. Among the major goals of cowpea breeding and improvement programs are the rapid manipulation of agronomic traits for seed size and quality and improved resistance to abiotic and biotic stresses to enhance productivity. Knowing the suite of transcription factors (TFs) and transcriptionally active proteins (TAPs) that control various critical plant cellular processes would contribute tremendously to these improvement aims.
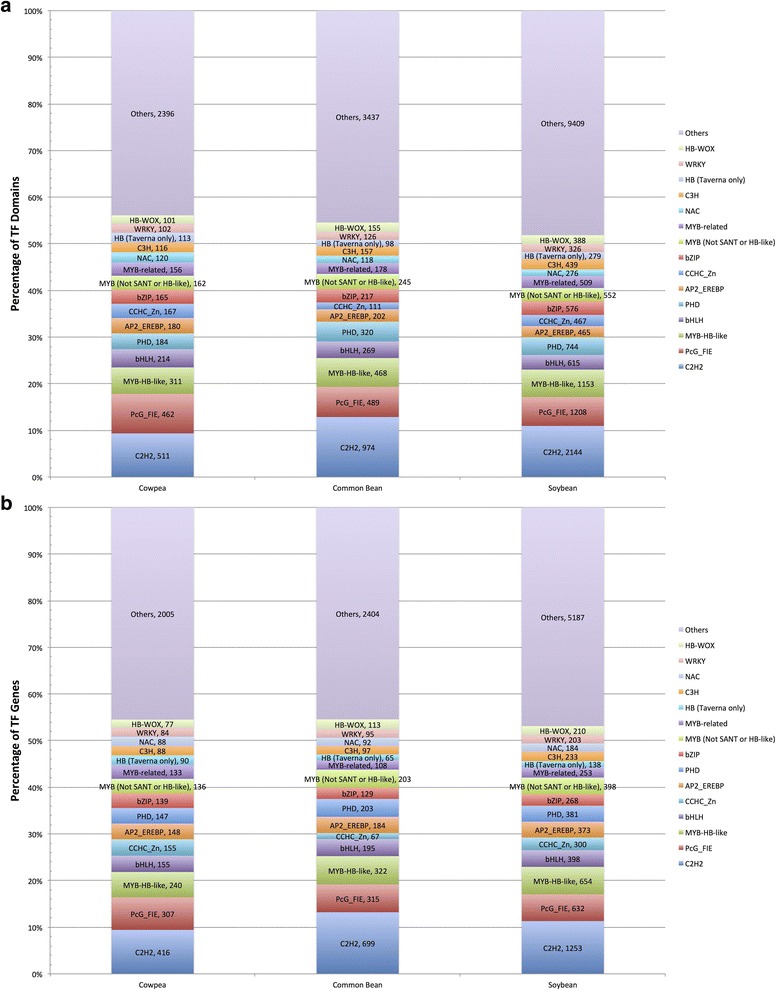
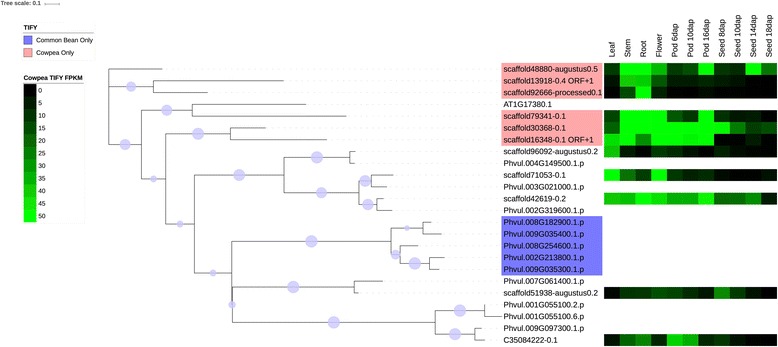
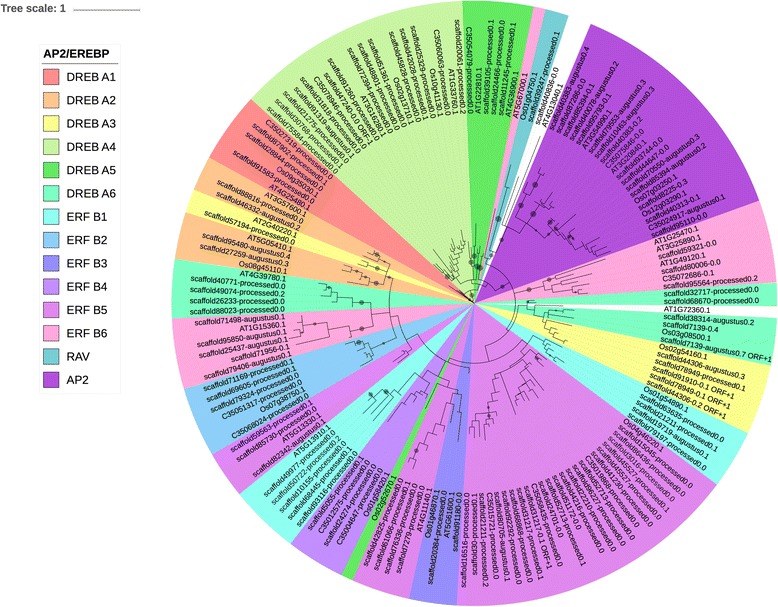
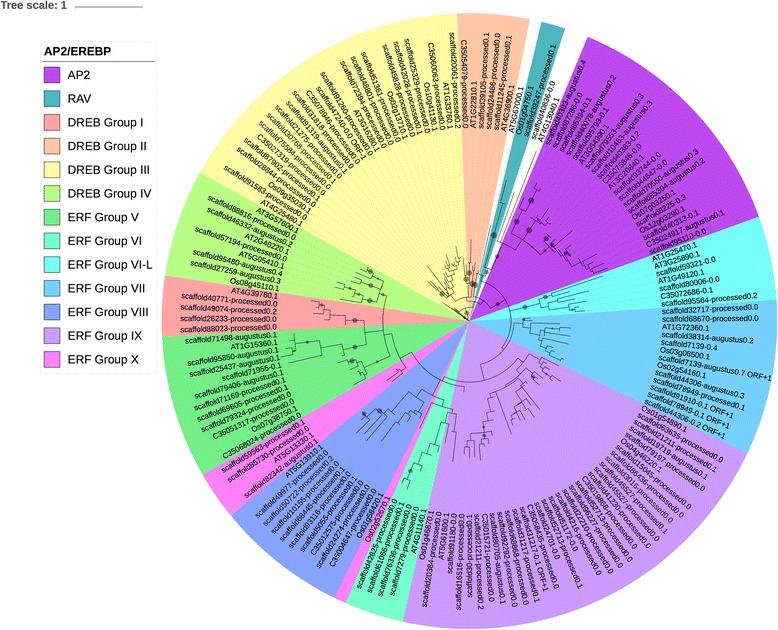
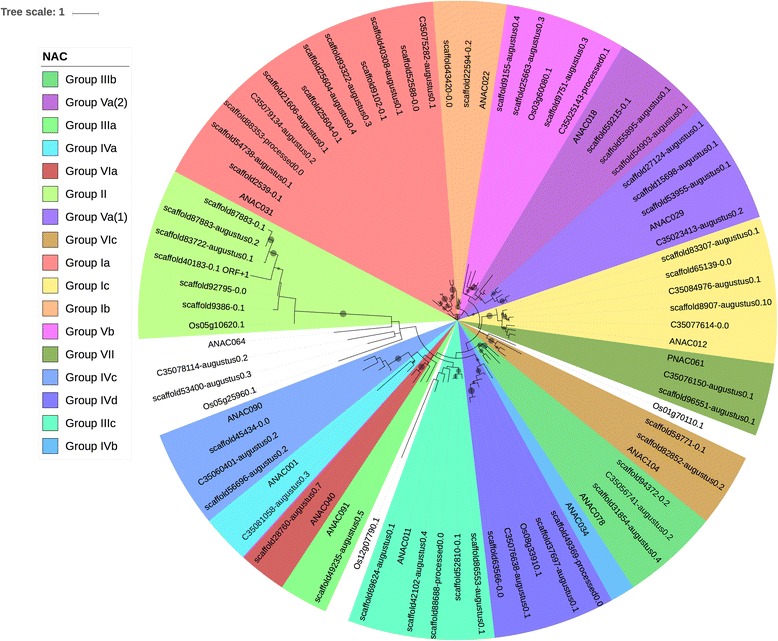
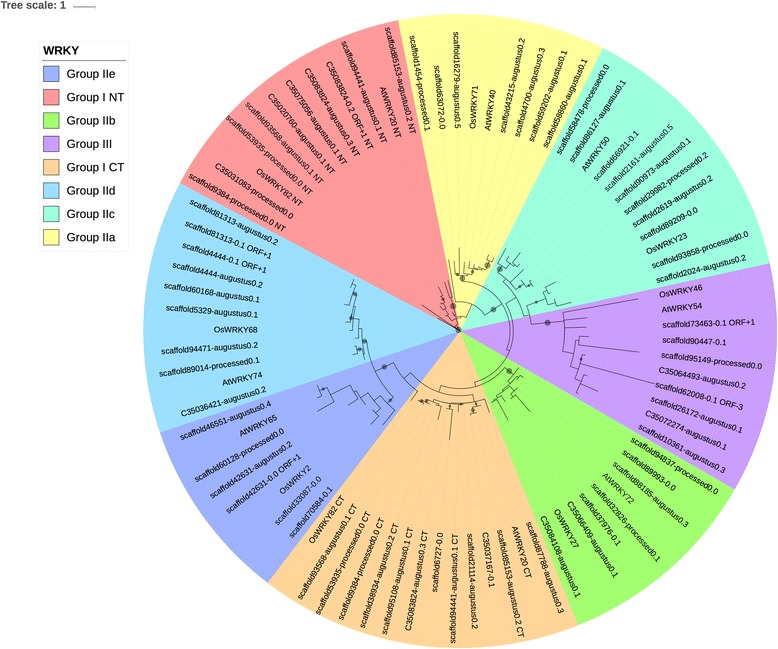
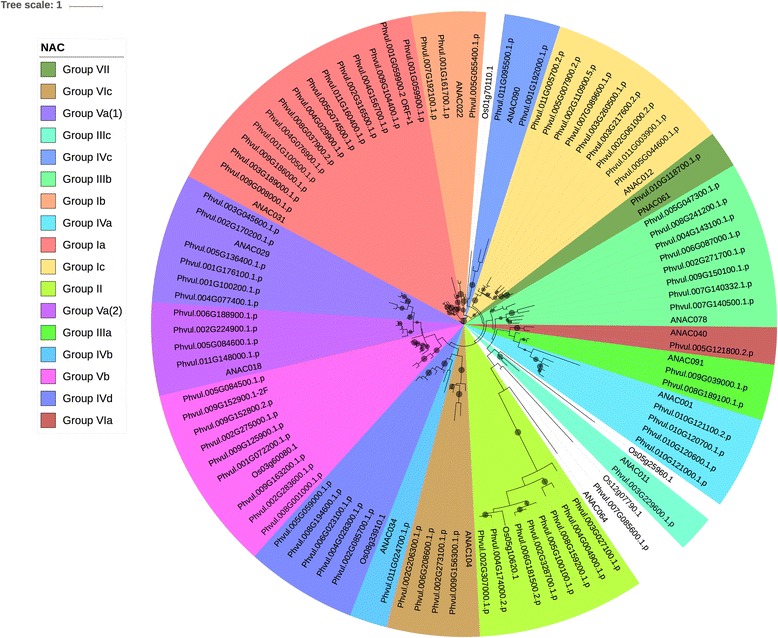
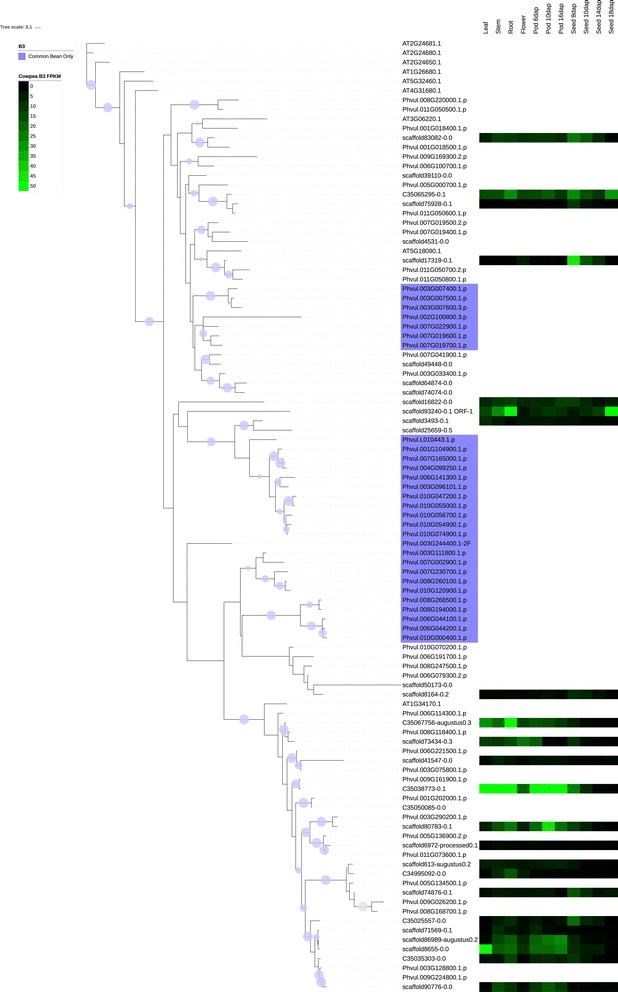
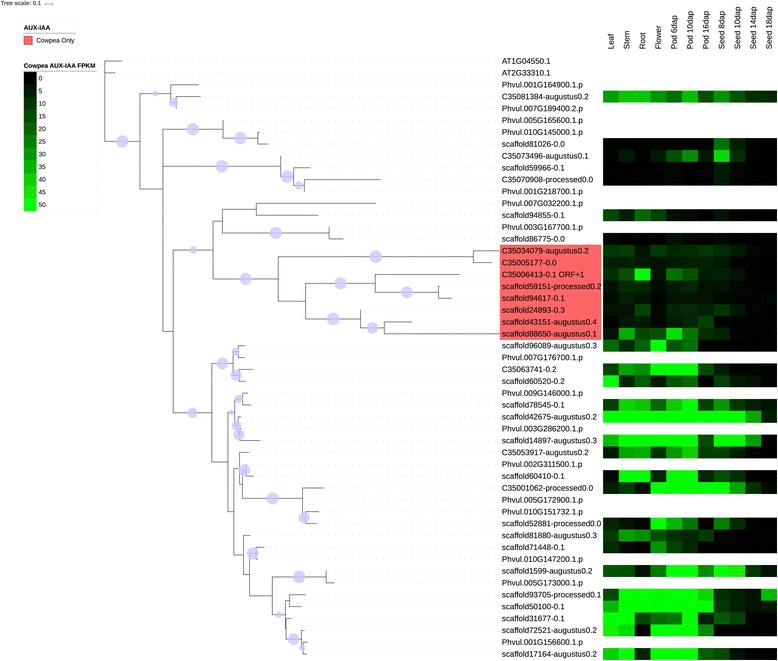
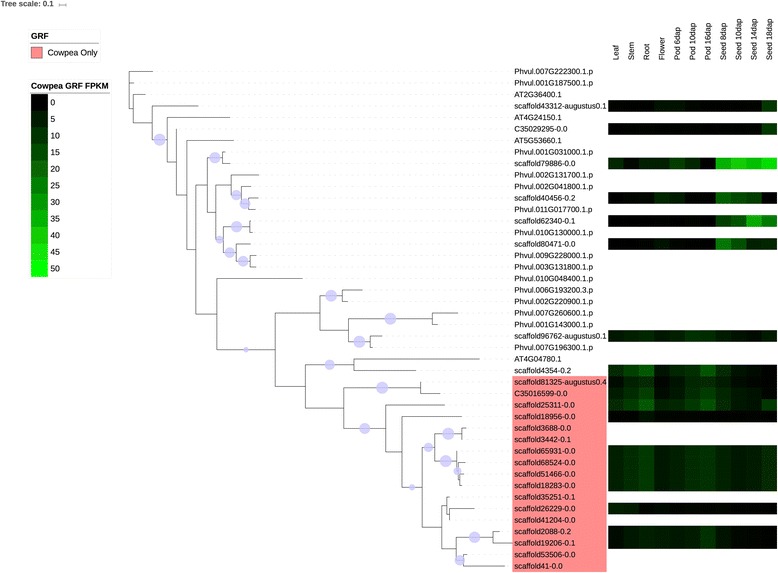
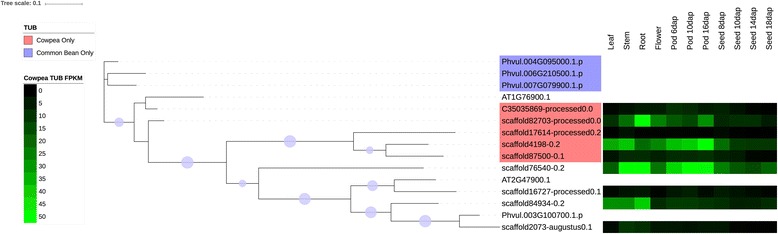
We used a computational approach that employed three different predictive pipelines to data mine the cowpea genome and identified over 4400 genes representing 136 different TF and TAP families. We compare the information content of cowpea to two evolutionarily close species common bean (Phaseolus vulgaris), and soybean (Glycine max) to gauge the relative informational content. Our data indicate that correcting for genome size cowpea has fewer TF and TAP genes than common bean (4408 / 5291) and soybean (4408/ 11,065). Members of the GROWTH-REGULATING FACTOR (GRF) and Auxin/indole-3-acetic acid (Aux/IAA) gene families appear to be over-represented in the genome relative to common bean and soybean, whereas members of the MADS (Minichromosome maintenance deficient 1 (MCM1), AGAMOUS, DEFICIENS, and serum response factor (SRF)) and C2C2-YABBY appear to be under-represented. Analysis of the AP2-EREBP APETALA2-Ethylene Responsive Element Binding Protein (AP2-EREBP), NAC (NAM (no apical meristem), ATAF1, 2 (Arabidopsis transcription activation factor), CUC (cup-shaped cotyledon)), and WRKY families, known to be important in defense signaling, revealed changes and phylogenetic rearrangements relative to common bean and soybean that suggest these groups may have evolved different functions.
The availability of detailed information on the coding capacity of the cowpea genome and in particular the various TF and TAP gene families will facilitate future comparative analysis and development of strategies for controlling growth, differentiation, and abiotic and biotic stress resistances of cowpea.
豇豆(Vignaunguiculata(L.)Walp.)是撒哈拉以南非洲半干旱热带地区最重要的粮食和饲料豆科植物,全球约 80%的产量主要来自低投入的自给农场所。豇豆育种和改良计划的主要目标之一是快速操纵农艺性状,包括种子大小和质量,并提高对非生物和生物胁迫的抗性,以提高生产力。了解控制各种关键植物细胞过程的转录因子(TF)和转录活性蛋白(TAP)的套件将极大地有助于实现这些改进目标。
我们使用了一种计算方法,该方法使用三种不同的预测管道对豇豆基因组进行数据挖掘,鉴定了超过 4400 个基因,代表 136 种不同的 TF 和 TAP 家族。我们将豇豆的信息含量与两种进化上相近的物种——普通菜豆(Phaseolus vulgaris)和大豆(Glycine max)进行比较,以衡量相对信息含量。我们的数据表明,校正基因组大小后,豇豆的 TF 和 TAP 基因比普通菜豆(4408/5291)和大豆(4408/11,065)少。生长调节因子(GRF)和生长素/吲哚-3-乙酸(Aux/IAA)基因家族的成员似乎在基因组中相对于普通菜豆和大豆过度表达,而 MADS(Minichromosome maintenance deficient 1(MCM1)、AGAMOUS、DEFICIENS 和血清反应因子(SRF))和 C2C2-YABBY 的成员则表达不足。对 AP2-EREBPAPETALA2-乙烯响应元件结合蛋白(AP2-EREBP)、NAC(NAM(无顶端分生组织)、ATAF1、2(拟南芥转录激活因子)、CUC(杯状子叶))和 WRKY 家族的分析表明,这些家族在防御信号转导中很重要,与普通菜豆和大豆相比,发生了变化和系统发育重排,表明这些家族可能具有不同的功能。
详细了解豇豆基因组的编码能力,特别是各种 TF 和 TAP 基因家族的信息,将有助于未来对豇豆生长、分化以及非生物和生物胁迫抗性的比较分析和控制策略的制定。