Weihs Karen L, Murphy William, Abbas Richat, Chiles Deborah, England Richard D, Ramaker Sara, Wajsbrot Dalia B
1 Department of Psychiatry, University of Arizona , Tucson, Arizona.
2 Psychiatric Associates , Overland Park, Kansas.
J Child Adolesc Psychopharmacol. 2018 Feb;28(1):36-46. doi: 10.1089/cap.2017.0100. Epub 2017 Nov 30.
To evaluate the short-term efficacy and safety of desvenlafaxine (25-50 mg/d) compared with placebo in children and adolescents with major depressive disorder (MDD).
Outpatient children (7-11 years) and adolescents (12-17 years) who met DSM-IV-TR criteria for MDD and had screening and baseline Children's Depression Rating Scale-Revised (CDRS-R) total scores >40 were randomly assigned to 8-week treatment with placebo, desvenlafaxine (25, 35, or 50 mg/d based on baseline weight), or fluoxetine (20 mg/d). The primary efficacy endpoint was change from baseline in CDRS-R total score at week 8, analyzed using a mixed-effects model for repeated measures. Secondary efficacy endpoints included week 8 Clinical Global Impressions-Severity, Clinical Global Impressions-Improvement (CGI-I), and response (CGI-I ≤ 2). Safety assessments included adverse events, physical and vital sign measurements, laboratory evaluations, electrocardiogram, and the Columbia-Suicide Severity Rating Scale.
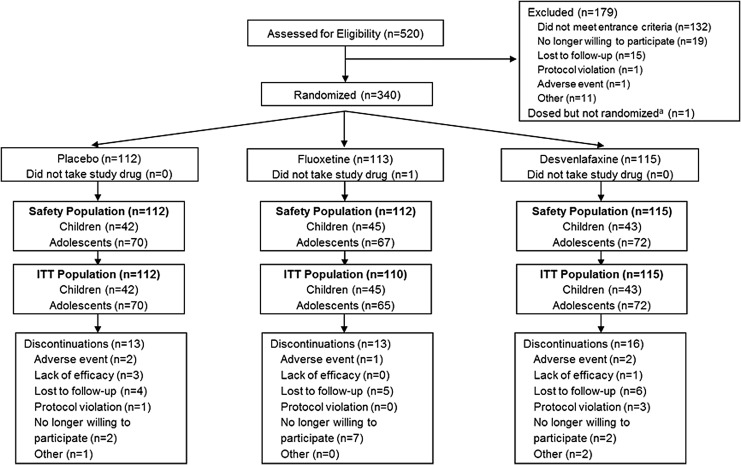
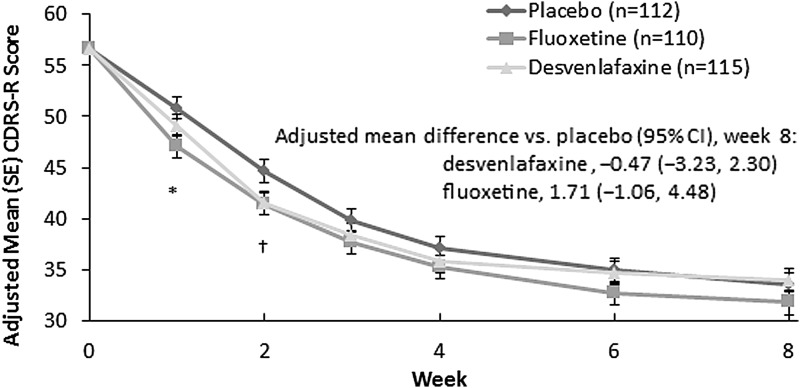
The safety population included 339 patients (children, n = 130; adolescents, n = 209). The primary endpoint, change from baseline in CDRS-R total score at week 8, did not statistically separate from placebo, for either desvenlafaxine (adjusted mean [standard error] change, -22.6 [1.17]) or fluoxetine (-24.8 [1.17]; placebo, -23.1 [1.18]). Week 8 CGI-I response rates were significantly greater for fluoxetine (78.2%; p = 0.017) than for placebo (62.6%); desvenlafaxine (68.7%) did not differ from placebo. Other secondary outcomes were consistent with those obtained with CDRS-R. Rates of treatment-emergent adverse events were comparable among treatment groups (desvenlafaxine, 60.0%; placebo, 70.5%; and fluoxetine, 64.3%).
Desvenlafaxine did not demonstrate efficacy for treating MDD in children and adolescents in this trial. Because neither desvenlafaxine nor the reference medication, fluoxetine, demonstrated a statistically significant difference from placebo on the primary endpoint, this was considered a failed trial and no efficacy conclusions can be drawn. Desvenlafaxine 25-50 mg/d was generally safe and well tolerated in children and adolescents in this study.
评估去甲文拉法辛(25 - 50毫克/天)与安慰剂相比,治疗儿童和青少年重度抑郁症(MDD)的短期疗效和安全性。
符合DSM - IV - TR标准的门诊儿童(7 - 11岁)和青少年(12 - 17岁),且筛查和基线儿童抑郁评定量表修订版(CDRS - R)总分>40,被随机分配接受为期8周的安慰剂、去甲文拉法辛(根据基线体重分别为25、35或50毫克/天)或氟西汀(20毫克/天)治疗。主要疗效终点是第8周时CDRS - R总分相对于基线的变化,采用重复测量的混合效应模型进行分析。次要疗效终点包括第8周的临床总体印象 - 严重程度、临床总体印象 - 改善情况(CGI - I)以及缓解情况(CGI - I≤2)。安全性评估包括不良事件、体格和生命体征测量、实验室检查、心电图以及哥伦比亚自杀严重程度评定量表。
安全人群包括339名患者(儿童,n = 130;青少年,n = 209)。主要终点,即第8周时CDRS - R总分相对于基线的变化,去甲文拉法辛(校正均值[标准误]变化,-22.6[1.17])或氟西汀(-24.8[1.17];安慰剂,-23.1[1.18])与安慰剂相比均无统计学差异。第8周时,氟西汀的CGI - I缓解率(78.2%;p = 0.017)显著高于安慰剂(62.6%);去甲文拉法辛(68.7%)与安慰剂无差异。其他次要结局与CDRS - R得出的结果一致。各治疗组中治疗中出现的不良事件发生率相当(去甲文拉法辛,60.0%;安慰剂,70.5%;氟西汀,64.3%)。
在本试验中,去甲文拉法辛未显示出治疗儿童和青少年MDD的疗效。由于去甲文拉法辛和对照药物氟西汀在主要终点上与安慰剂均无统计学显著差异,因此本试验被认为失败,无法得出疗效结论。本研究中,25 - 50毫克/天的去甲文拉法辛在儿童和青少年中总体安全且耐受性良好。