Shell Jasmine, Patel Dhaval, Powers Astin, Quezado Martha, Killian Keith, Meltzer Paul, Zhu Jack, Gaitanidis Apostolos, Karzai Fatima, Neychev Vladimir, Green Patience, Kebebew Electron
Endocrine Oncology Branch, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892.
Laboratory of Pathology, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892.
J Endocr Soc. 2017 Jul 7;1(9):1124-1134. doi: 10.1210/js.2017-00156. eCollection 2017 Sep 1.
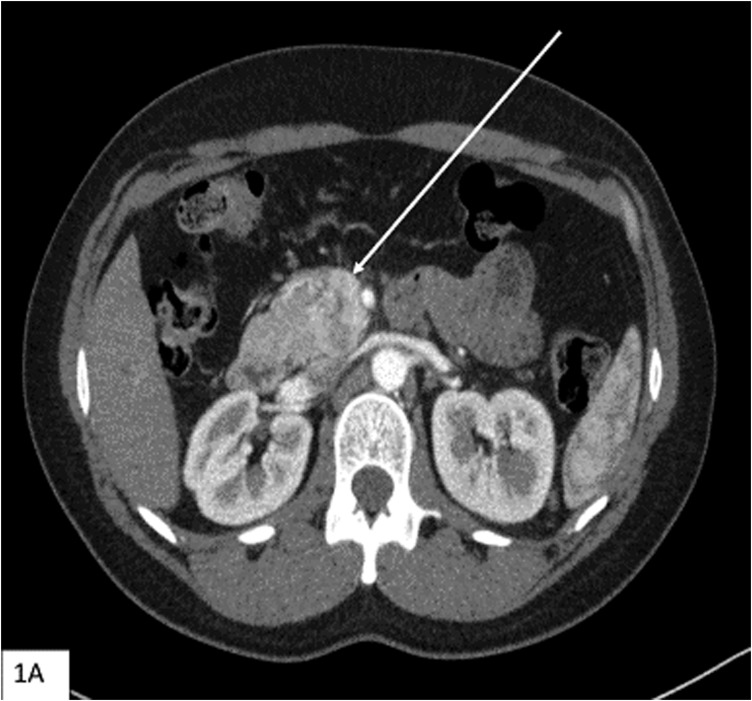
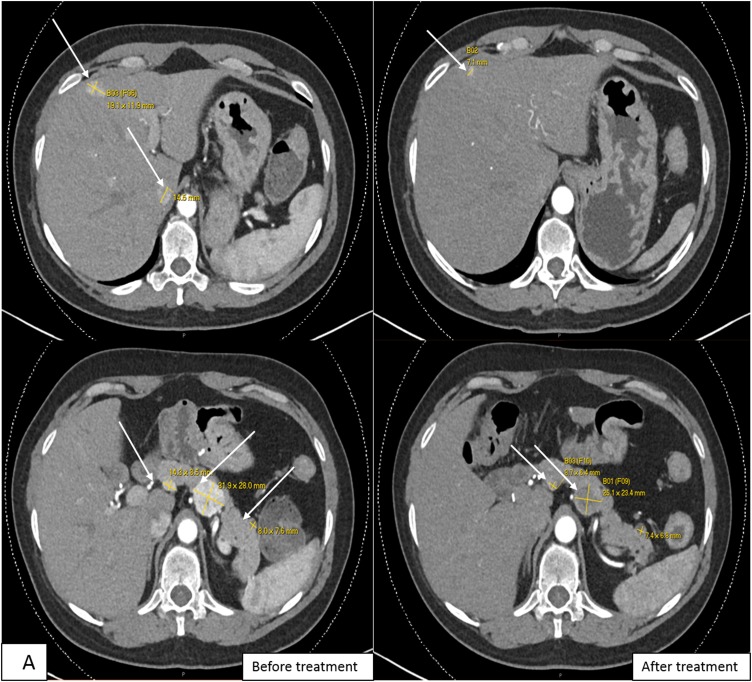
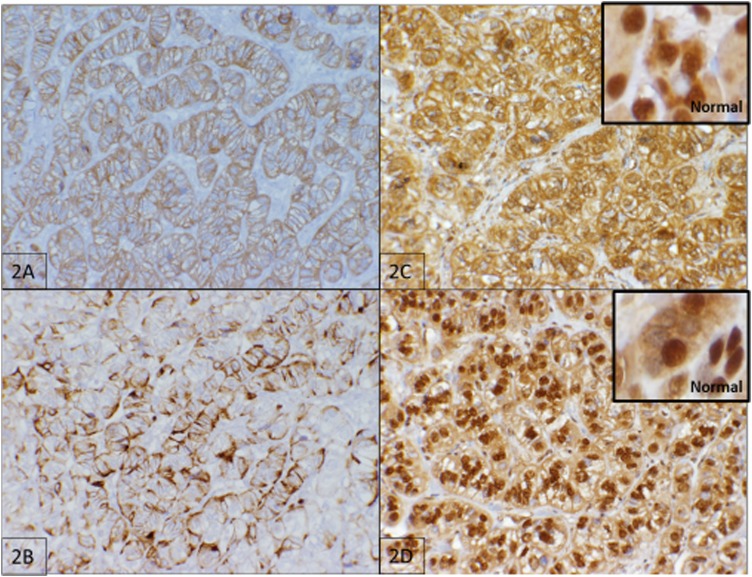
Multiple endocrine neoplasia type 1 (MEN1) and von Hippel-Lindau (VHL) are autosomal-dominant diseases caused by germline mutations in tumor-suppressor genes. A patient with a germline mutation and a somatic mutation in the tumor has not been reported. Herein, we report on a patient with MEN1 and a metastatic nonfunctioning pancreatic neuroendocrine tumor (PNET) with a somatic mutation. This patient underwent a pancreaticoduodenectomy for a grade 2 PNET obstructing her pancreatic duct. The patient developed liver and regional lymph node metastases as well as growth of a PNET in the remnant pancreas. As part of a clinical trial for mutation-targeted therapy, a biopsy of the metastatic tumor was obtained. The clinical diagnosis, confirmed by OncoVAR-NET and molecular profiling analysis, revealed MEN1 with a germline deletion in exon 2 and a c.402 deletion C, p.Phe134LeufsX51. In addition, a somatic mutation in the gene-a nonsense mutation, c.529A>T, p.Arg177Ter-was identified by hybrid capture sequencing. The mutations were confirmed by Sanger sequencing. Comparative genomic hybridization showed loss of heterozygosity in both the MEN1 and VHL genes. The patient was treated with sunitinib and had a partial response to treatment. This case illustrates not only that a second hit occurs in tumor suppressor genes but that somatic mutations are also possible in additional tumor suppressor genes. This suggests that targeted therapy selection should include analysis of somatic mutations even when the susceptibility gene is known.
多发性内分泌腺瘤1型(MEN1)和冯希佩尔-林道病(VHL)是由肿瘤抑制基因的种系突变引起的常染色体显性疾病。尚未有肿瘤中存在种系突变和体细胞突变的患者的报道。在此,我们报告一名患有MEN1和转移性无功能胰腺神经内分泌肿瘤(PNET)且存在体细胞突变的患者。该患者因2级PNET阻塞胰管而接受了胰十二指肠切除术。患者出现了肝和区域淋巴结转移,以及残余胰腺中PNET的生长。作为针对突变的靶向治疗临床试验的一部分,获取了转移性肿瘤的活检样本。经OncoVAR-NET和分子谱分析证实的临床诊断显示,该患者患有MEN1,外显子2存在种系缺失,以及c.402缺失C,p.Phe134LeufsX51。此外,通过杂交捕获测序鉴定出该基因存在体细胞突变——一个无义突变,c.529A>T,p.Arg177Ter。这些突变通过桑格测序得到证实。比较基因组杂交显示MEN1和VHL基因均存在杂合性缺失。该患者接受了舒尼替尼治疗,对治疗有部分反应。这个病例不仅说明了肿瘤抑制基因会发生第二次打击,而且额外的肿瘤抑制基因也可能发生体细胞突变。这表明即使已知易感基因,靶向治疗的选择也应包括对体细胞突变的分析。