Biochemistry Interdisciplinary Graduate Program, Molecular and Cellular Biochemistry Department, Indiana University, Bloomington, IN, USA.
Turku Centre for Biotechnology, University of Turku and Åbo Academy University, Turku, Finland.
Pain. 2018 May;159(5):849-863. doi: 10.1097/j.pain.0000000000001152.
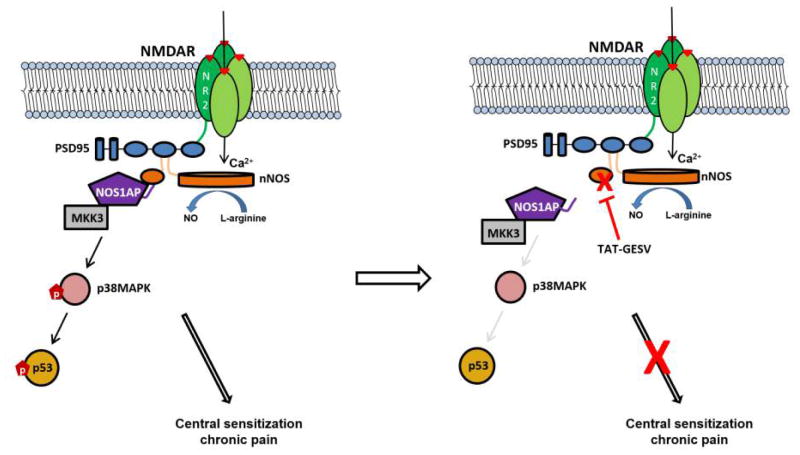
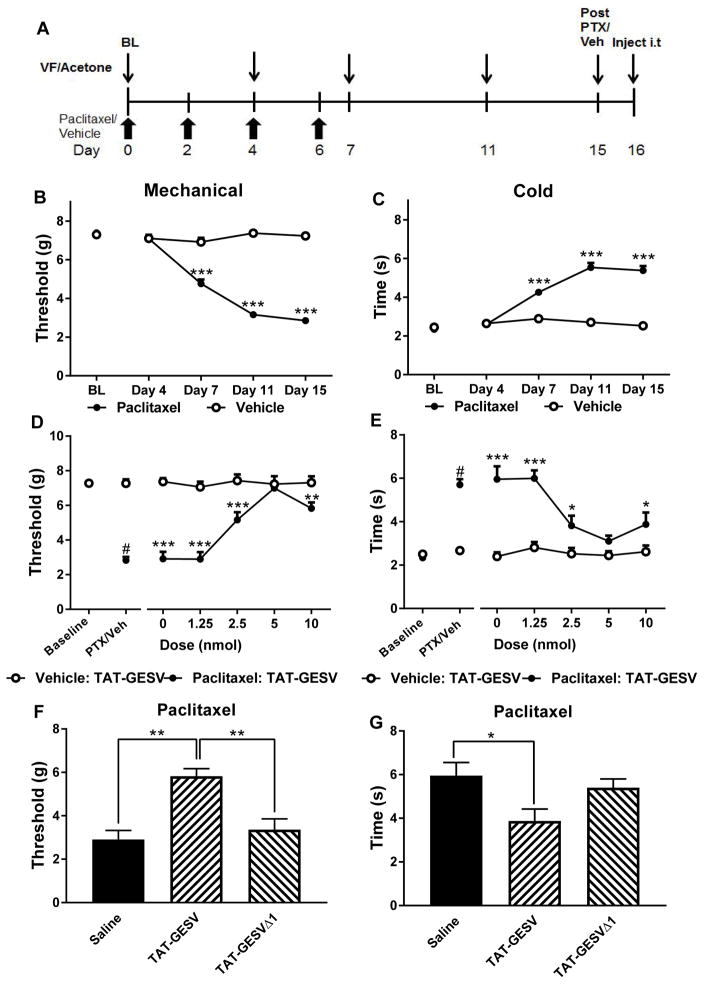
Elevated N-methyl-D-aspartate receptor (NMDAR) activity is linked to central sensitization and chronic pain. However, NMDAR antagonists display limited therapeutic potential because of their adverse side effects. Novel approaches targeting the NR2B-PSD95-nNOS complex to disrupt signaling pathways downstream of NMDARs show efficacy in preclinical pain models. Here, we evaluated the involvement of interactions between neuronal nitric oxide synthase (nNOS) and the nitric oxide synthase 1 adaptor protein (NOS1AP) in pronociceptive signaling and neuropathic pain. TAT-GESV, a peptide inhibitor of the nNOS-NOS1AP complex, disrupted the in vitro binding between nNOS and its downstream protein partner NOS1AP but not its upstream protein partner postsynaptic density 95 kDa (PSD95). Putative inactive peptides (TAT-cp4GESV and TAT-GESVΔ1) failed to do so. Only the active peptide protected primary cortical neurons from glutamate/glycine-induced excitotoxicity. TAT-GESV, administered intrathecally (i.t.), suppressed mechanical and cold allodynia induced by either the chemotherapeutic agent paclitaxel or a traumatic nerve injury induced by partial sciatic nerve ligation. TAT-GESV also blocked the paclitaxel-induced phosphorylation at Ser15 of p53, a substrate of p38 MAPK. Finally, TAT-GESV (i.t.) did not induce NMDAR-mediated motor ataxia in the rotarod test and did not alter basal nociceptive thresholds in the radiant heat tail-flick test. These observations support the hypothesis that antiallodynic efficacy of an nNOS-NOS1AP disruptor may result, at least in part, from blockade of p38 MAPK-mediated downstream effects. Our studies demonstrate, for the first time, that disrupting nNOS-NOS1AP protein-protein interactions attenuates mechanistically distinct forms of neuropathic pain without unwanted motor ataxic effects of NMDAR antagonists.
N-甲基-D-天冬氨酸受体(NMDAR)活性升高与中枢敏化和慢性疼痛有关。然而,由于其不良反应,NMDAR 拮抗剂的治疗潜力有限。靶向 NR2B-PSD95-nNOS 复合物的新型方法可破坏 NMDAR 下游信号通路,在临床前疼痛模型中显示出疗效。在这里,我们评估了神经元型一氧化氮合酶(nNOS)与一氧化氮合酶 1 衔接蛋白(NOS1AP)之间相互作用在伤害性信号转导和神经病理性疼痛中的参与情况。TAT-GESV 是 nNOS-NOS1AP 复合物的肽抑制剂,可破坏 nNOS 与其下游蛋白伴侣 NOS1AP 之间的体外结合,但不破坏其上游蛋白伴侣突触后密度 95kDa(PSD95)。假定的无活性肽(TAT-cp4GESV 和 TAT-GESVΔ1)则无法做到这一点。只有活性肽可保护原代皮质神经元免受谷氨酸/甘氨酸诱导的兴奋性毒性。鞘内给予 TAT-GESV 可抑制紫杉醇引起的机械性和冷性痛觉过敏,以及部分坐骨神经结扎引起的创伤性神经损伤。TAT-GESV 还可阻断紫杉醇诱导的 p53 的丝氨酸 15 磷酸化,p53 是 p38 MAPK 的底物。最后,鞘内给予 TAT-GESV 不会引起旋转棒试验中的 NMDAR 介导的运动性共济失调,也不会改变辐射热尾触试验中的基础痛觉阈值。这些观察结果支持这样一种假设,即 nNOS-NOS1AP 破坏剂的抗痛觉过敏疗效至少部分源自对 p38 MAPK 介导的下游效应的阻断。我们的研究首次表明,破坏 nNOS-NOS1AP 蛋白-蛋白相互作用可减轻机制上不同形式的神经病理性疼痛,而不会产生 NMDAR 拮抗剂的不必要的运动性共济失调作用。