Department of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, PA, USA.
Department of Pathology, University of Colorado, Denver, CO, USA.
Mod Pathol. 2018 May;31(5):690-704. doi: 10.1038/modpathol.2017.182. Epub 2018 Jan 12.
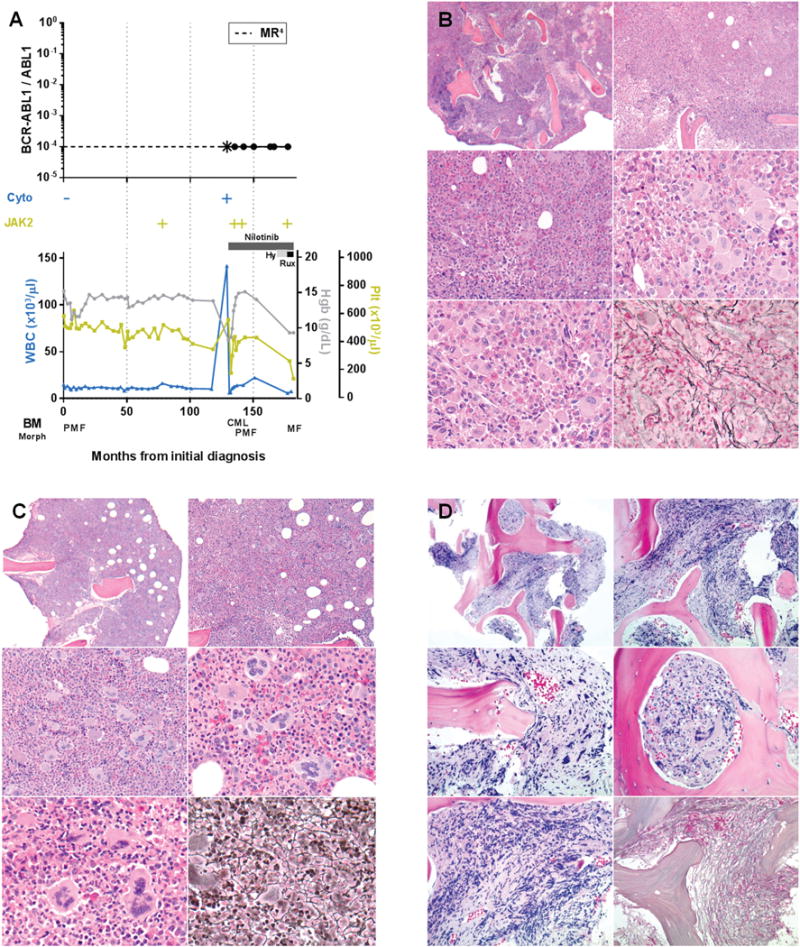
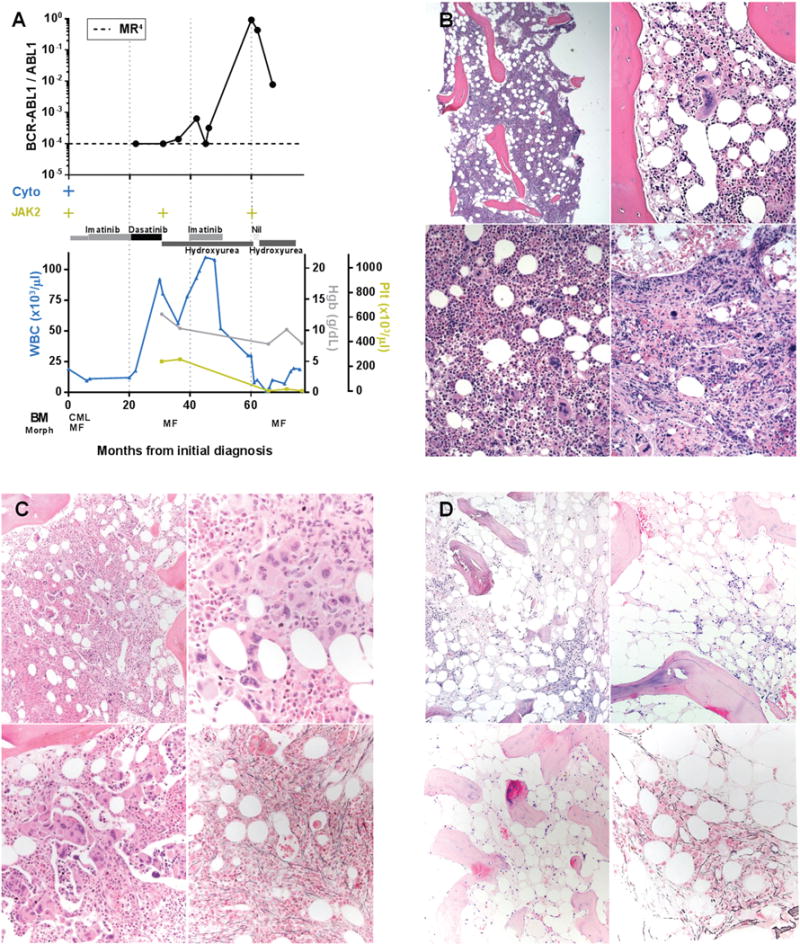
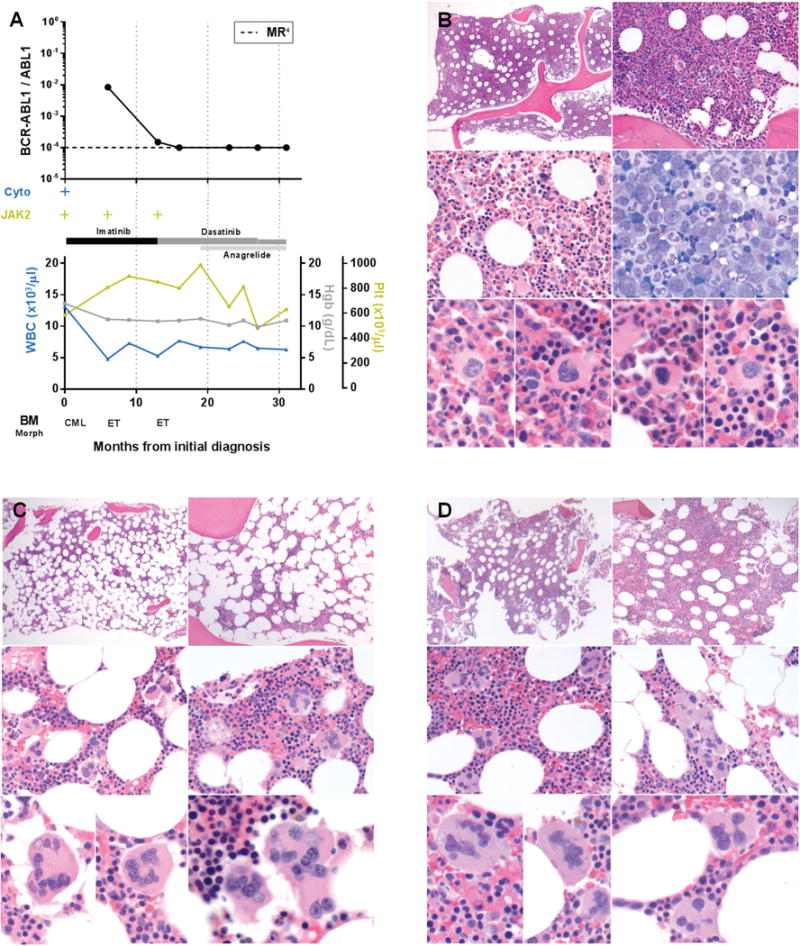
Myeloproliferative neoplasms arise from hematopoietic stem cells with somatically altered tyrosine kinase signaling. Classification of myeloproliferative neoplasms is based on hematologic, histopathologic and molecular characteristics including the presence of the BCR-ABL1 and JAK2 V617F. Although thought to be mutually exclusive, a number of cases with co-occurring BCR-ABL1 and JAK2 V617F have been identified. To characterize the clinicopathologic features of myeloproliferative neoplasms with concomitant BCR-ABL1 and JAK2 V617F, and define the frequency of co-occurrence, we conducted a retrospective multi-institutional study. Cases were identified using a search of electronic databases over a decade at six major institutions. Of 1570 patients who were tested for both BCR-ABL1 and JAK2 V617F, six were positive for both. An additional five patients were identified via clinical records providing a total of 11 cases for detailed evaluation. For each case, clinical variables, hematologic and genetic data, and bone marrow histomorphologic features were analyzed. The sequence of identification of the genetic abnormalities varied: five patients were initially diagnosed with a JAK2 V617F+ myeloproliferative neoplasm, one patient initially had BCR-ABL1+ chronic myeloid leukemia, while both alterations were identified simultaneously in five patients. Classification of the BCR-ABL1-negative myeloproliferative neoplasms varied, and in some cases, features only became apparent following tyrosine kinase inhibitor therapy. Seven of the 11 patients showed myelofibrosis, in some cases before identification of the second genetic alteration. Our data, reflecting the largest reported study comprehensively detailing clinicopathologic features and response to therapy, show that the co-occurrence of BCR-ABL1 and JAK2 V617F is rare, with an estimated frequency of 0.4%, and most often reflects two distinct ('composite') myeloproliferative neoplasms. Although uncommon, it is important to be aware of this potentially confounding genetic combination, lest these features be misinterpreted to reflect resistance to therapy or disease progression, considerations that could lead to inappropriate management.
骨髓增殖性肿瘤起源于具有体细胞改变的酪氨酸激酶信号的造血干细胞。骨髓增殖性肿瘤的分类基于血液学、组织病理学和分子特征,包括 BCR-ABL1 和 JAK2 V617F 的存在。尽管认为它们是相互排斥的,但已经发现了一些同时存在 BCR-ABL1 和 JAK2 V617F 的病例。为了描述同时存在 BCR-ABL1 和 JAK2 V617F 的骨髓增殖性肿瘤的临床病理特征,并确定其共存频率,我们进行了一项回顾性多机构研究。在六个主要机构的十多年电子数据库搜索中确定了病例。在接受 BCR-ABL1 和 JAK2 V617F 检测的 1570 名患者中,有 6 名患者同时检测出两种异常。通过临床记录又发现了 5 例患者,总共 11 例患者进行了详细评估。对于每个病例,分析了临床变量、血液学和遗传学数据以及骨髓组织形态学特征。基因异常的识别顺序不同:5 例患者最初被诊断为 JAK2 V617F+骨髓增殖性肿瘤,1 例患者最初患有 BCR-ABL1+慢性髓性白血病,而 5 例患者同时发现了两种改变。BCR-ABL1 阴性骨髓增殖性肿瘤的分类不同,在某些情况下,仅在酪氨酸激酶抑制剂治疗后才出现特征。11 例患者中有 7 例出现骨髓纤维化,在某些情况下,在确定第二种遗传改变之前就出现了。我们的数据反映了最大的报告研究,全面详细地描述了临床病理特征和对治疗的反应,表明 BCR-ABL1 和 JAK2 V617F 的共存非常罕见,估计频率为 0.4%,并且最常反映两种不同的(“复合”)骨髓增殖性肿瘤。虽然罕见,但重要的是要意识到这种潜在的混淆性遗传组合,以免这些特征被误解为反映对治疗的耐药或疾病进展,这可能导致不适当的管理。