Vervloessem Tamara, Akl Haidar, Tousseyn Thomas, De Smedt Humbert, Parys Jan B, Bultynck Geert
KU Leuven, Laboratory of Molecular and Cellular Signaling, Department of Cellular and Molecular Medicine & Leuven Kanker Instituut (LKI), Leuven, Belgium.
Current/Present address: Lebanese University, Department of Biology, Hadath, Lebanon.
Oncotarget. 2017 Dec 4;8(67):111656-111671. doi: 10.18632/oncotarget.22898. eCollection 2017 Dec 19.
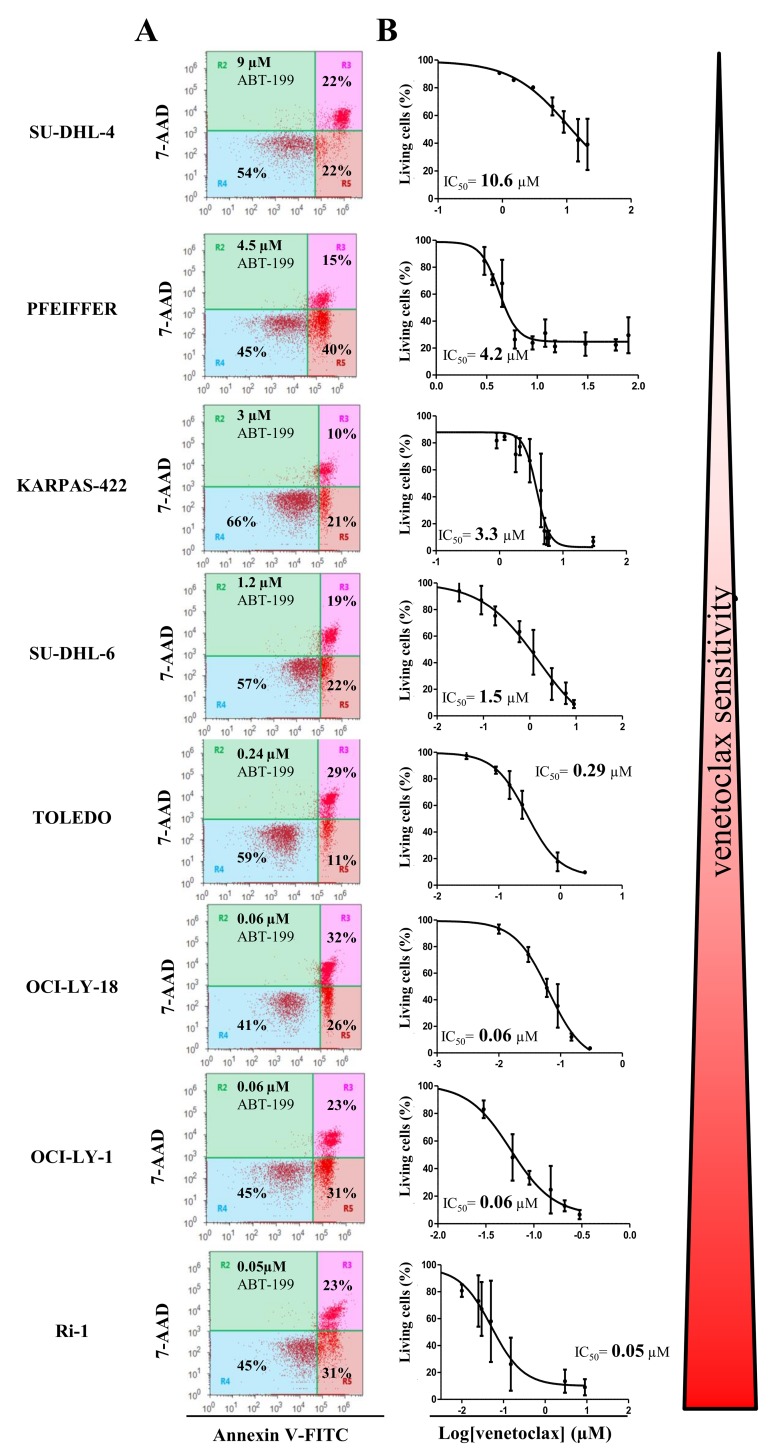
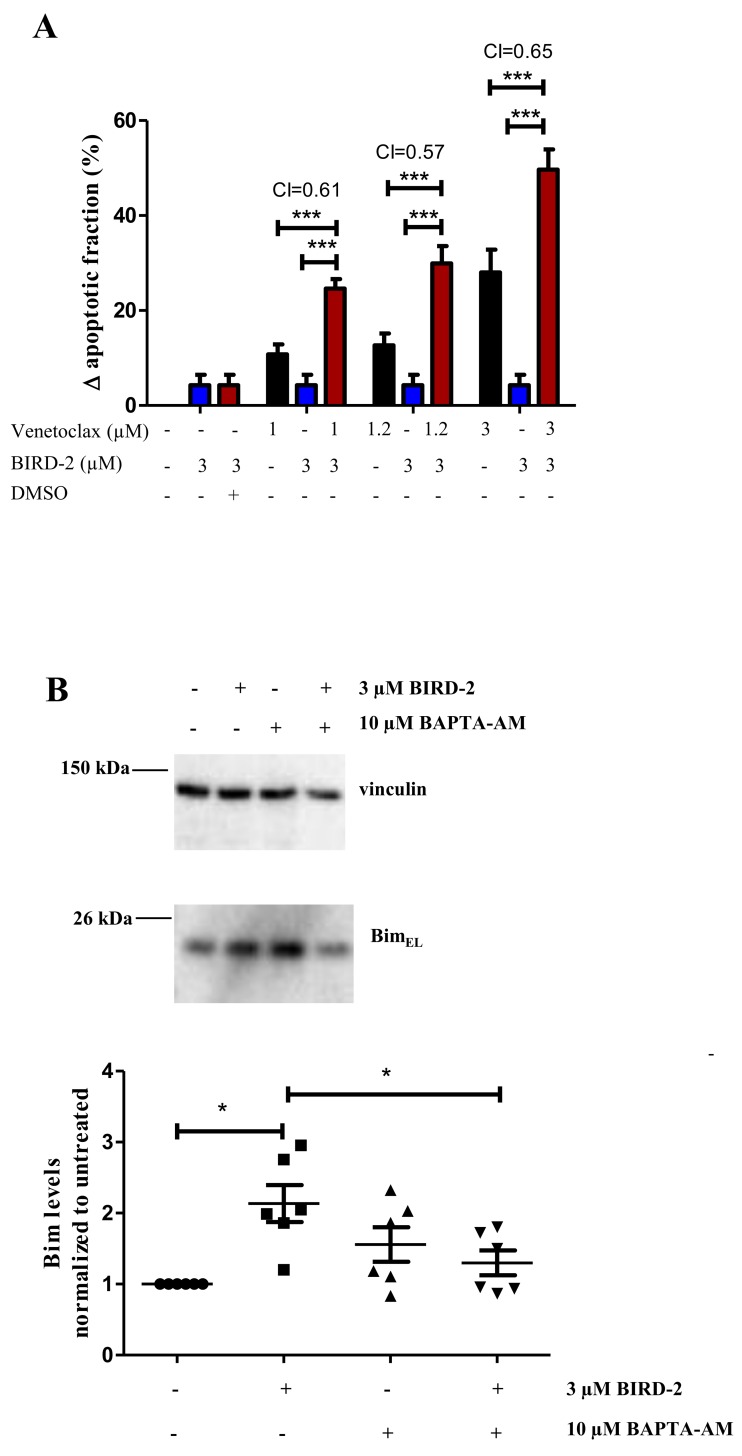
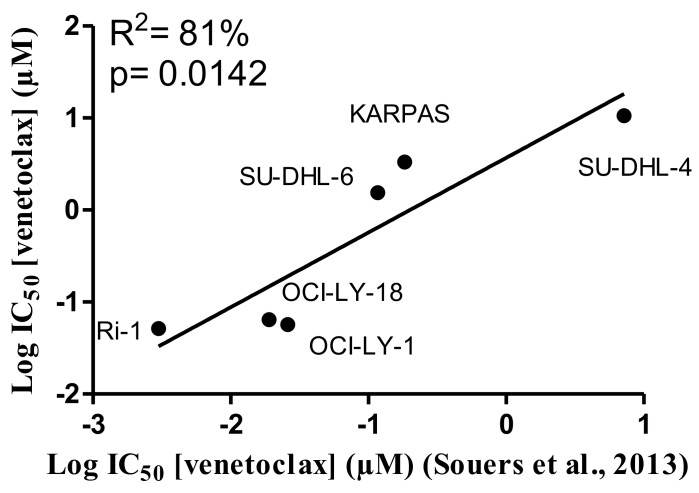
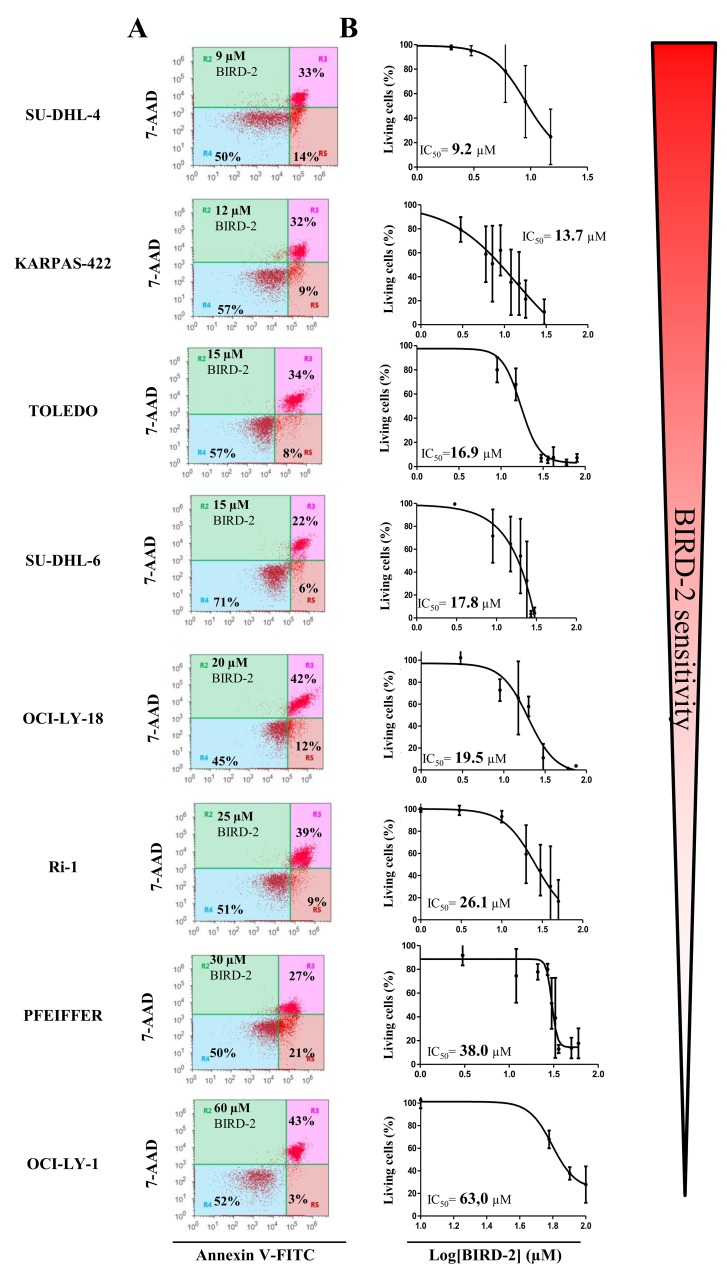
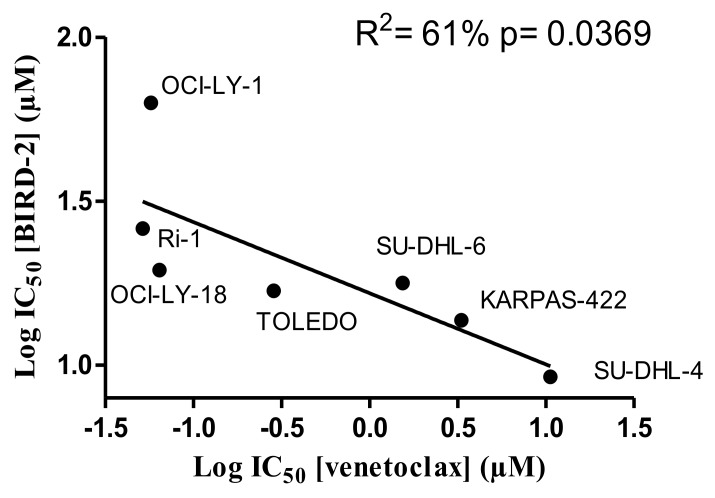
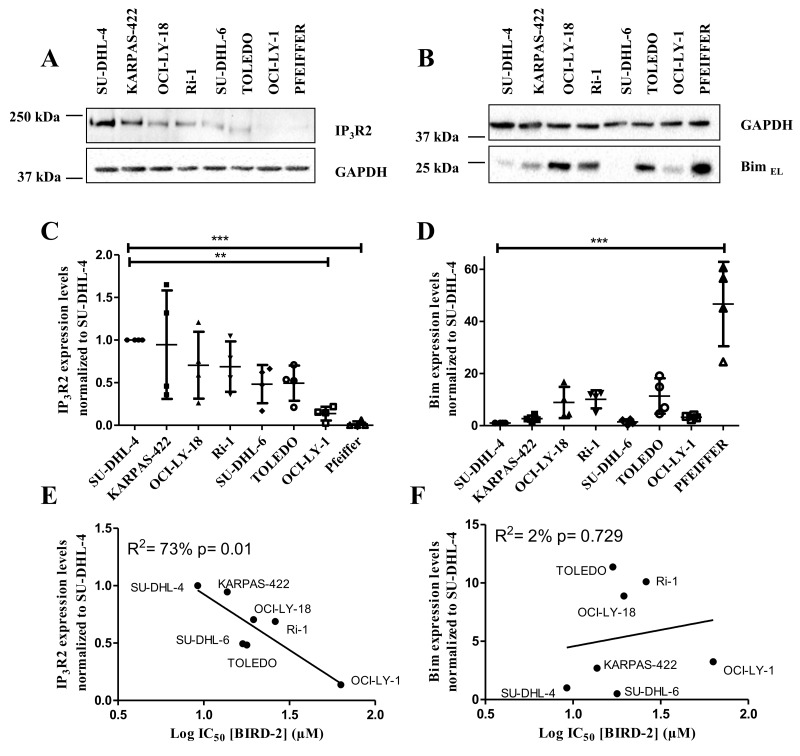
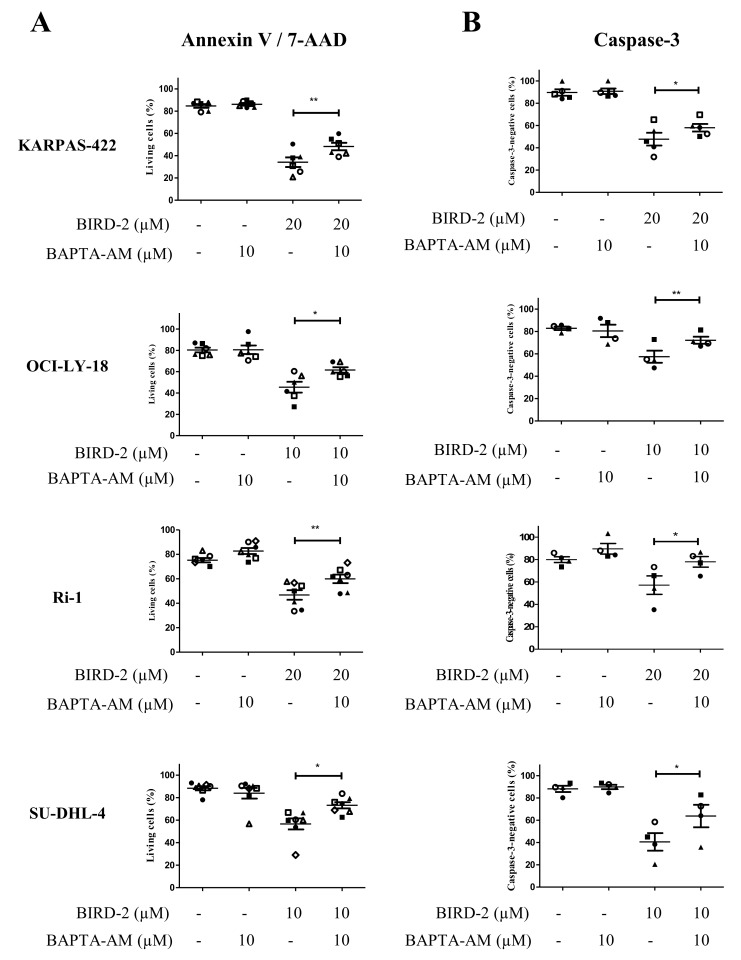
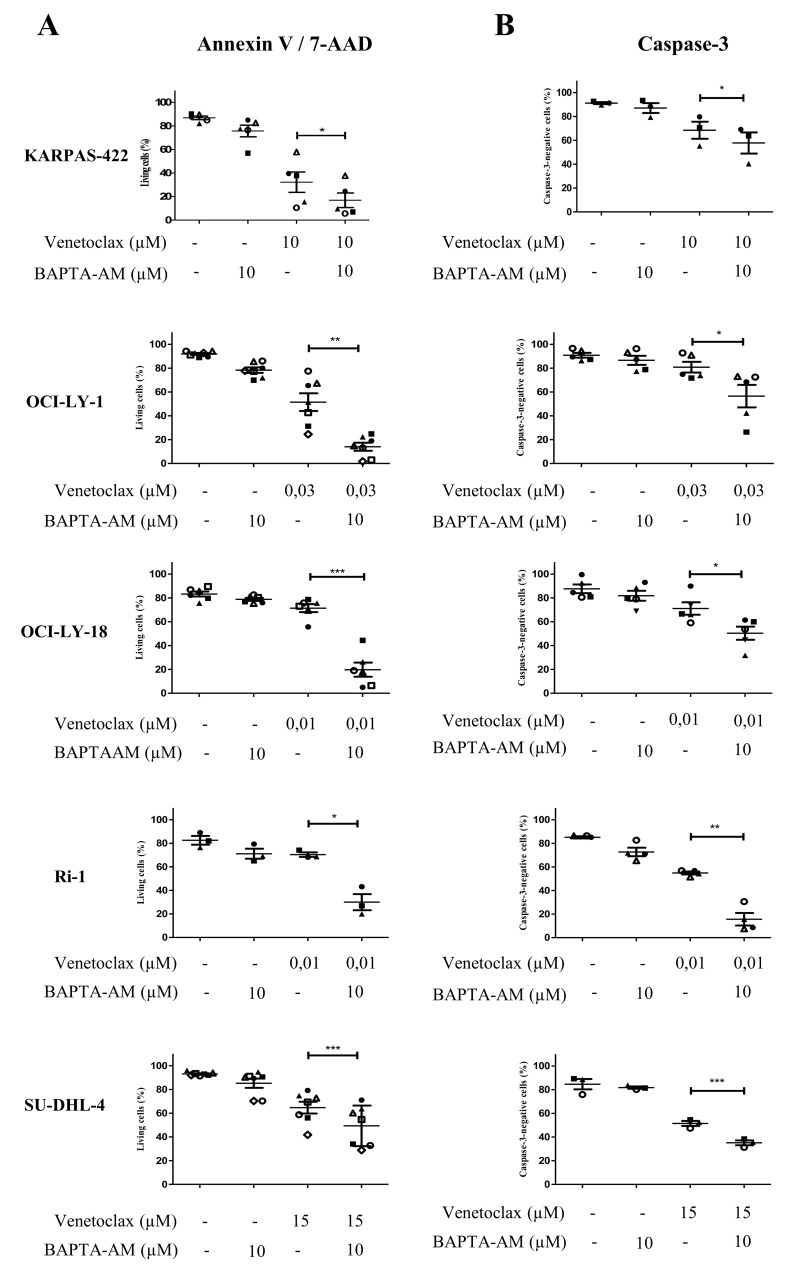
Bcl-2 is often upregulated in cancers to neutralize the BH3-only protein Bim at the mitochondria. BH3 mimetics (e.g. ABT-199 (venetoclax)) kill cancer cells by targeting Bcl-2's hydrophobic cleft and disrupting Bcl-2/Bim complexes. Some cancers with elevated Bcl-2 display poor responses towards BH3 mimetics, suggesting an additional function for anti-apoptotic Bcl-2 in these cancers. Indeed, Bcl-2 via its BH4 domain prevents cytotoxic Ca release from the endoplasmic reticulum (ER) by directly inhibiting the inositol 1,4,5-trisphosphate receptor (IPR). The cell-permeable Bcl-2/IPR disruptor-2 (BIRD-2) peptide can kill these Bcl-2-dependent cancers by targeting Bcl-2's BH4 domain, unleashing pro-apoptotic Ca-release events. We compared eight "primed to death" diffuse large B-cell lymphoma cell lines (DLBCL) for their apoptotic sensitivity towards BIRD-2 and venetoclax. By determining their IC using cytometric cell-death analysis, we discovered a reciprocal sensitivity towards venetoclax versus BIRD-2. Using immunoblotting, we quantified the expression levels of IPR2 and Bim in DLBCL cell lysates, revealing that BIRD-2 sensitivity correlated with IPR2 levels but not with Bim levels. Moreover, the requirement of intracellular Ca for BIRD-2- venetoclax-induced cell death was different. Indeed, BAPTA-AM suppressed BIRD-2-induced cell death, but promoted venetoclax-induced cell death in DLBCL cells. Finally, compared to single-agent treatments, combining BIRD-2 with venetoclax synergistically enhanced cell-death induction, correlating with a Ca-dependent upregulation of Bim after BIRD-2 treatment. Our findings suggest that some cancer cells require Bcl-2 proteins at the mitochondria, preventing Bax activation via its hydrophobic cleft, while others require Bcl-2 proteins at the ER, preventing cytotoxic Ca-signaling events via its BH4 domain.
Bcl-2在癌症中常常上调,以在线粒体中中和仅含BH3结构域的蛋白Bim。BH3模拟物(如ABT-199(维奈托克))通过靶向Bcl-2的疏水裂缝并破坏Bcl-2/Bim复合物来杀死癌细胞。一些Bcl-2升高的癌症对BH3模拟物反应不佳,这表明抗凋亡Bcl-2在这些癌症中具有额外功能。事实上,Bcl-2通过其BH4结构域直接抑制肌醇1,4,5-三磷酸受体(IPR),从而防止细胞毒性钙从内质网(ER)释放。细胞可渗透的Bcl-2/IPR破坏剂-2(BIRD-2)肽可通过靶向Bcl-2的BH4结构域杀死这些依赖Bcl-2的癌症,引发促凋亡的钙释放事件。我们比较了8种“濒死”弥漫性大B细胞淋巴瘤(DLBCL)细胞系对BIRD-2和维奈托克的凋亡敏感性。通过使用细胞计数细胞死亡分析确定它们的半数抑制浓度(IC),我们发现了对维奈托克与BIRD-2的反向敏感性。使用免疫印迹法,我们定量了DLBCL细胞裂解物中IPR2和Bim的表达水平,发现对BIRD-2的敏感性与IPR2水平相关,但与Bim水平无关。此外,细胞内钙对BIRD-2和维奈托克诱导的细胞死亡需求不同。事实上,BAPTA-AM抑制BIRD-2诱导的细胞死亡,但促进DLBCL细胞中维奈托克诱导的细胞死亡。最后,与单药治疗相比,将BIRD-2与维奈托克联合使用可协同增强细胞死亡诱导,这与BIRD-2治疗后Bim的钙依赖性上调相关。我们的研究结果表明,一些癌细胞在线粒体需要Bcl-2蛋白,通过其疏水裂缝阻止Bax激活,而另一些癌细胞在内质网需要Bcl-2蛋白,通过其BH4结构域阻止细胞毒性钙信号事件。