Institut Curie, PSL Research University, Mines Paris Tech, INSERM U900, Paris 75005, France.
Département de Recherche Translationnelle, Institut Curie, PSL Research University, INSERM U830, Laboratoire RTOP (Recherche Translationelle en Oncologie Pédiatrique), SIREDO Oncology Center (Care, Innovation and research for children and AYA with cancer), Paris 75005, France.
Bioinformatics. 2018 Jun 1;34(11):1808-1816. doi: 10.1093/bioinformatics/bty016.
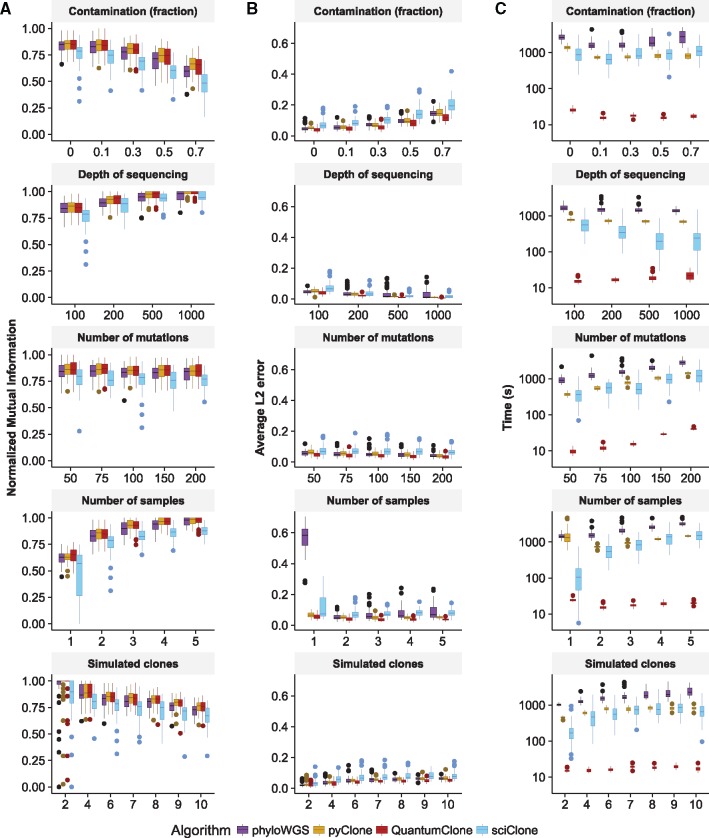
In cancer, clonal evolution is assessed based on information coming from single nucleotide variants and copy number alterations. Nonetheless, existing methods often fail to accurately combine information from both sources to truthfully reconstruct clonal populations in a given tumor sample or in a set of tumor samples coming from the same patient. Moreover, previously published methods detect clones from a single set of variants. As a result, compromises have to be done between stringent variant filtering [reducing dispersion in variant allele frequency estimates (VAFs)] and using all biologically relevant variants.
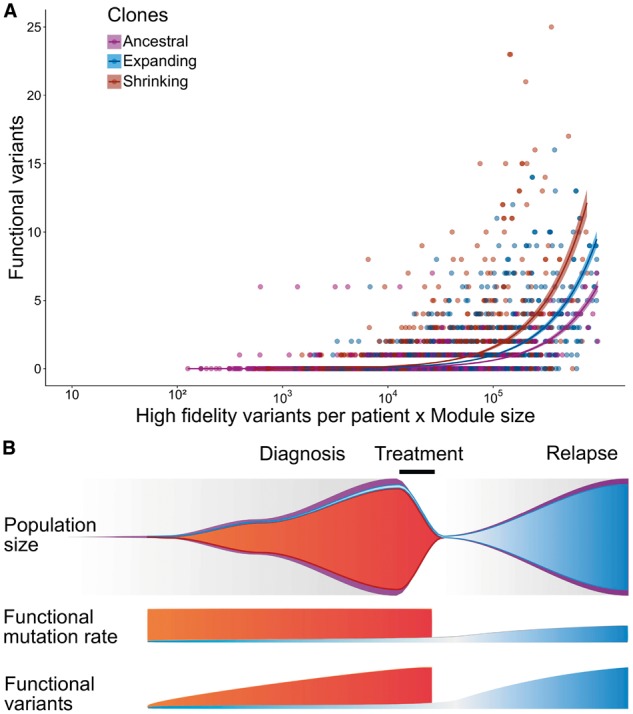
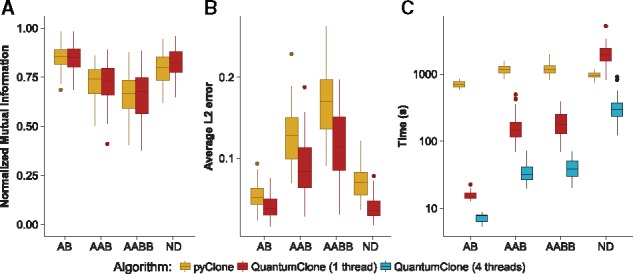
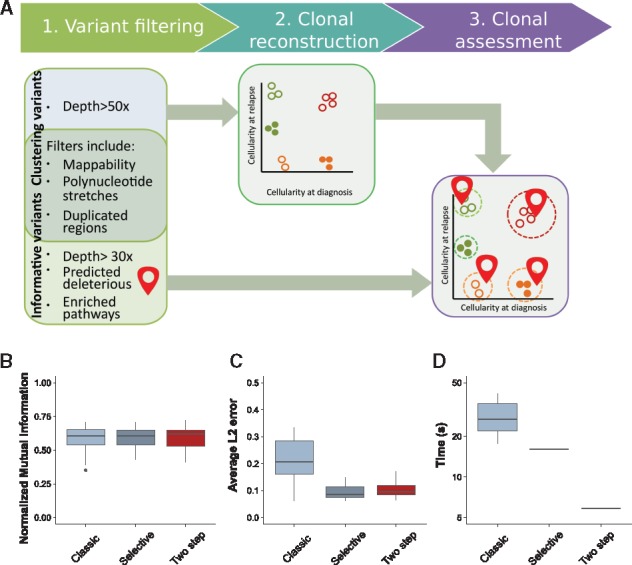
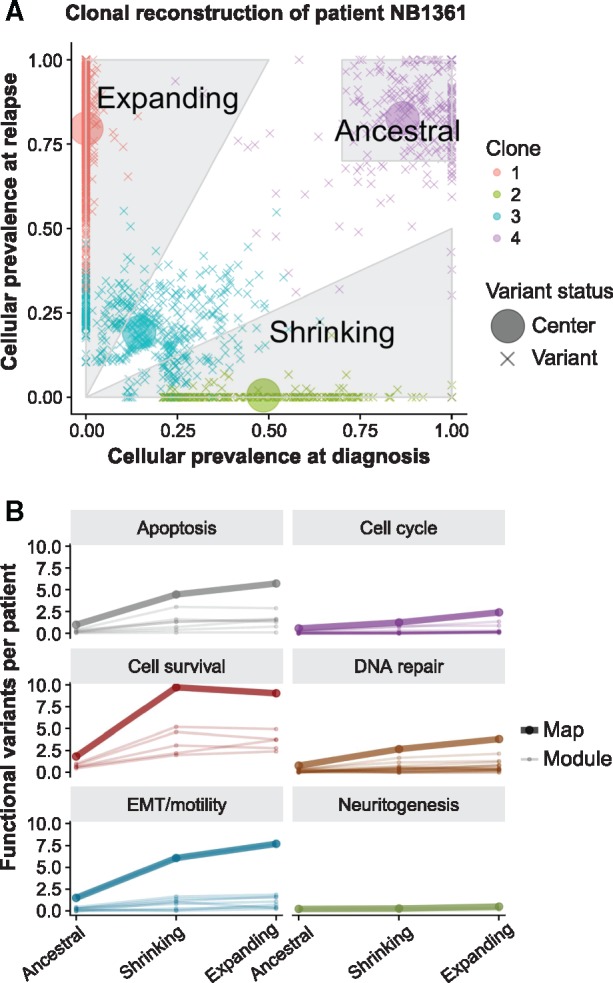
We present a framework for defining cancer clones using most reliable variants of high depth of coverage and assigning functional mutations to the detected clones. The key element of our framework is QuantumClone, a method for variant clustering into clones based on VAFs, genotypes of corresponding regions and information about tumor purity. We validated QuantumClone and our framework on simulated data. We then applied our framework to whole genome sequencing data for 19 neuroblastoma trios each including constitutional, diagnosis and relapse samples. We confirmed an enrichment of damaging variants within such pathways as MAPK (mitogen-activated protein kinases), neuritogenesis, epithelial-mesenchymal transition, cell survival and DNA repair. Most pathways had more damaging variants in the expanding clones compared to shrinking ones, which can be explained by the increased total number of variants between these two populations. Functional mutational rate varied for ancestral clones and clones shrinking or expanding upon treatment, suggesting changes in clone selection mechanisms at different time points of tumor evolution.
Source code and binaries of the QuantumClone R package are freely available for download at https://CRAN.R-project.org/package=QuantumClone.
gudrun.schleiermacher@curie.fr or valentina.boeva@inserm.fr.
Supplementary data are available at Bioinformatics online.
在癌症中,克隆进化是基于单核苷酸变体和拷贝数改变的信息来评估的。然而,现有的方法往往无法准确地结合两种来源的信息,以真实地重建给定肿瘤样本或来自同一患者的一组肿瘤样本中的克隆群体。此外,以前发表的方法从单一变异集检测克隆。因此,必须在严格的变异过滤(降低变异等位基因频率估计值 (VAF) 的离散度)和使用所有生物学相关的变异之间做出妥协。
我们提出了一个使用高深度覆盖的最可靠变异来定义癌症克隆并将功能突变分配给检测到的克隆的框架。我们框架的关键元素是 QuantumClone,这是一种基于 VAF、对应区域的基因型和肿瘤纯度信息将变异聚类为克隆的方法。我们在模拟数据上验证了 QuantumClone 和我们的框架。然后,我们将我们的框架应用于 19 个神经母细胞瘤三联体的全基因组测序数据,每个三联体包括正常、诊断和复发样本。我们证实了 MAPK(丝裂原活化蛋白激酶)、神经突发生、上皮-间充质转化、细胞存活和 DNA 修复等途径中的破坏性变异富集。与萎缩克隆相比,在扩张克隆中,大多数途径的破坏性变异更多,这可以用这两个群体之间变异总数的增加来解释。祖克隆和治疗后收缩或扩张的克隆的功能突变率不同,这表明在肿瘤进化的不同时间点,克隆选择机制发生了变化。
QuantumClone R 包的源代码和二进制文件可在 https://CRAN.R-project.org/package=QuantumClone 上免费下载。
gudrun.schleiermacher@curie.fr 或 valentina.boeva@inserm.fr。
补充数据可在 Bioinformatics 在线获取。