Miller Christopher A, White Brian S, Dees Nathan D, Griffith Malachi, Welch John S, Griffith Obi L, Vij Ravi, Tomasson Michael H, Graubert Timothy A, Walter Matthew J, Ellis Matthew J, Schierding William, DiPersio John F, Ley Timothy J, Mardis Elaine R, Wilson Richard K, Ding Li
The Genome Institute, Washington University, St. Louis, Missouri, United States of America.
The Genome Institute, Washington University, St. Louis, Missouri, United States of America; Department of Internal Medicine, Division of Oncology, Washington University School of Medicine, St. Louis, Missouri, United States of America.
PLoS Comput Biol. 2014 Aug 7;10(8):e1003665. doi: 10.1371/journal.pcbi.1003665. eCollection 2014 Aug.
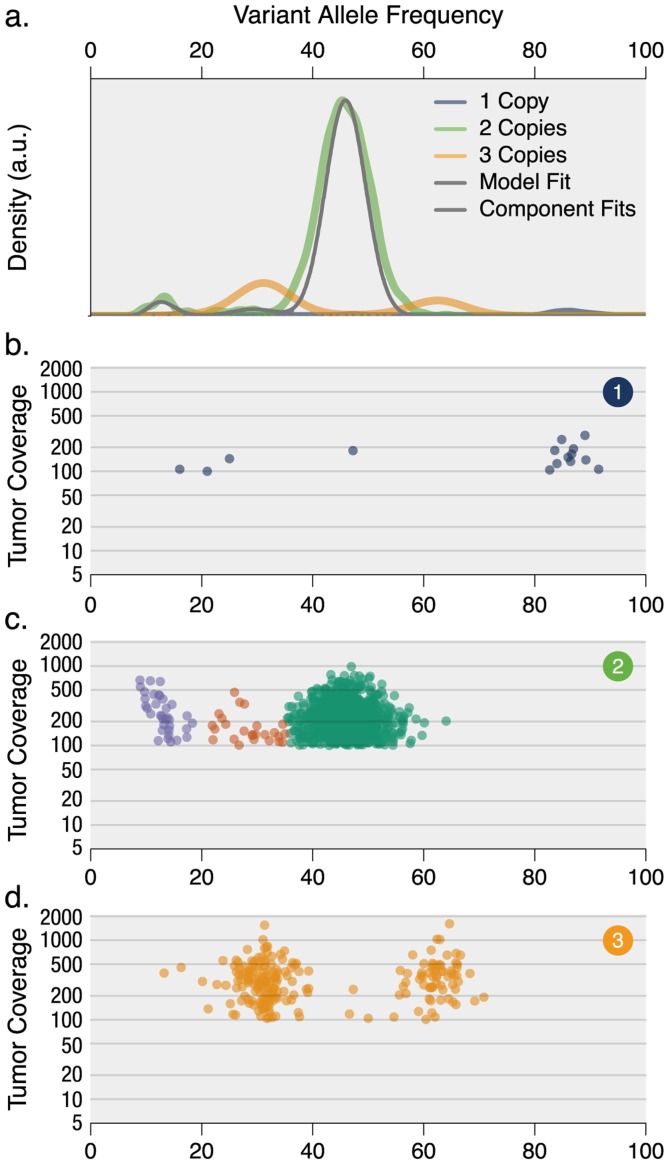
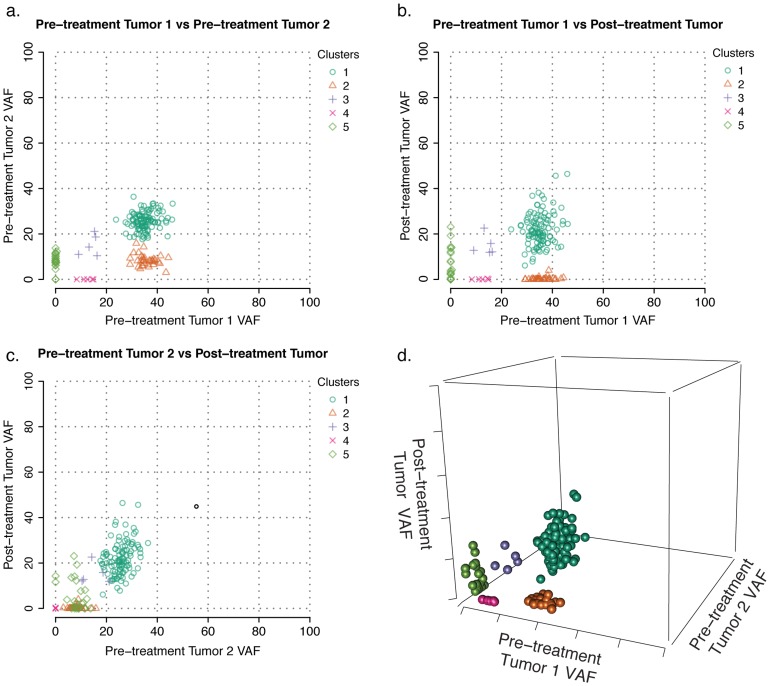
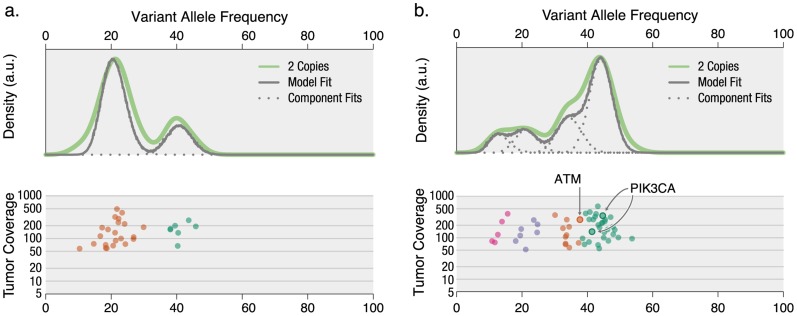
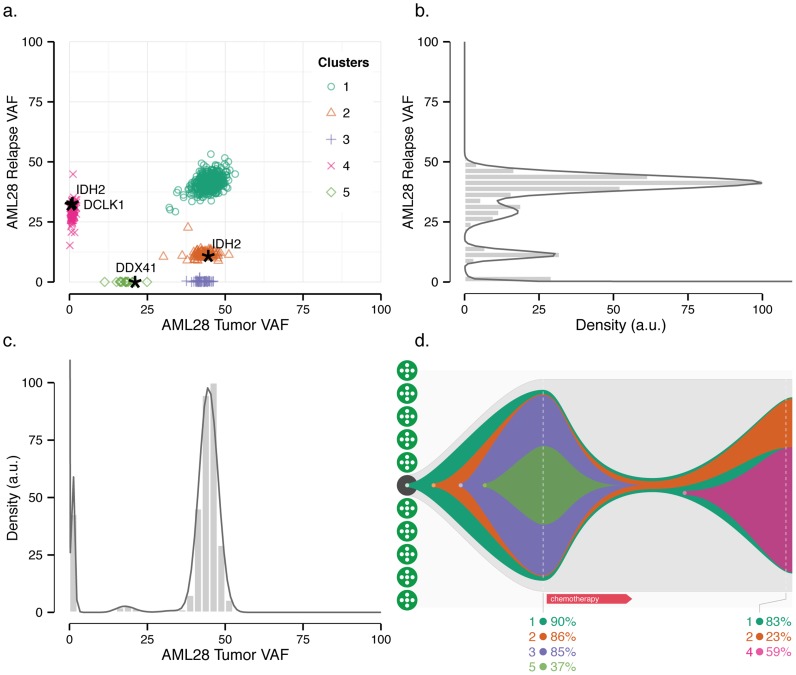
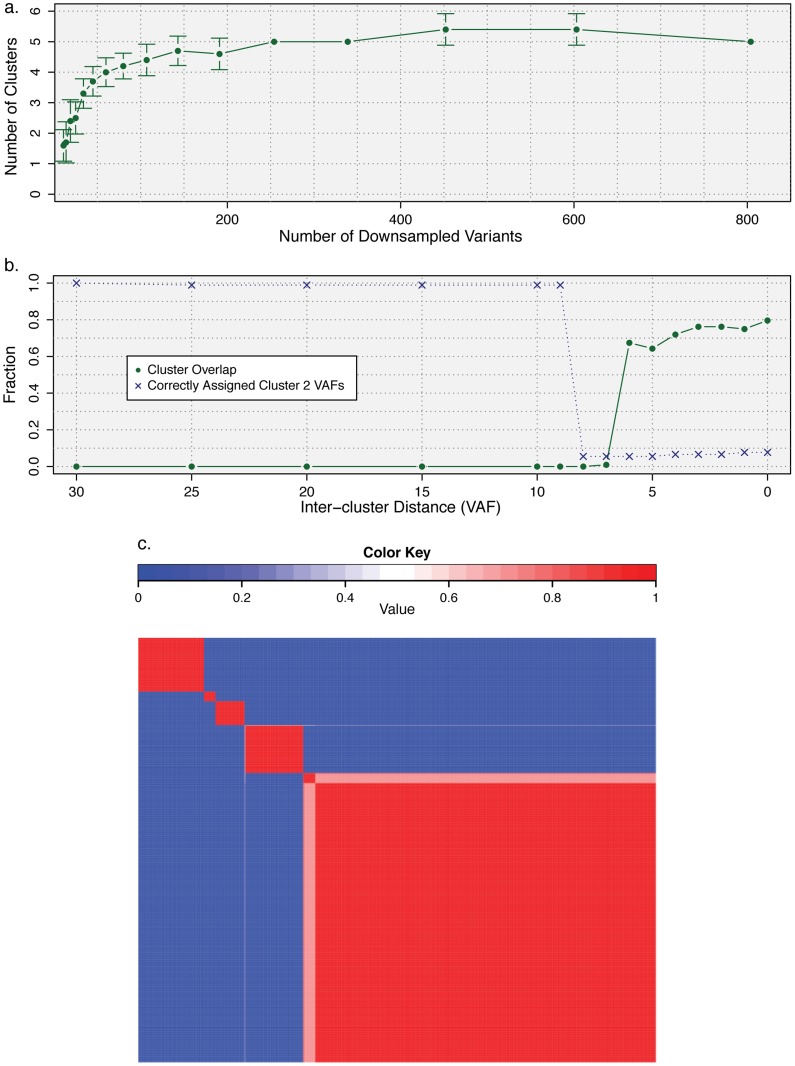
The sensitivity of massively-parallel sequencing has confirmed that most cancers are oligoclonal, with subpopulations of neoplastic cells harboring distinct mutations. A fine resolution view of this clonal architecture provides insight into tumor heterogeneity, evolution, and treatment response, all of which may have clinical implications. Single tumor analysis already contributes to understanding these phenomena. However, cryptic subclones are frequently revealed by additional patient samples (e.g., collected at relapse or following treatment), indicating that accurately characterizing a tumor requires analyzing multiple samples from the same patient. To address this need, we present SciClone, a computational method that identifies the number and genetic composition of subclones by analyzing the variant allele frequencies of somatic mutations. We use it to detect subclones in acute myeloid leukemia and breast cancer samples that, though present at disease onset, are not evident from a single primary tumor sample. By doing so, we can track tumor evolution and identify the spatial origins of cells resisting therapy.
大规模平行测序的敏感性已证实,大多数癌症是寡克隆性的,肿瘤细胞亚群具有不同的突变。这种克隆结构的精细解析有助于深入了解肿瘤异质性、进化和治疗反应,所有这些都可能具有临床意义。对单个肿瘤的分析已经有助于理解这些现象。然而,额外的患者样本(例如,在复发时或治疗后采集)经常会揭示出隐匿的亚克隆,这表明准确表征肿瘤需要分析同一患者的多个样本。为满足这一需求,我们提出了SciClone,这是一种通过分析体细胞突变的变异等位基因频率来识别亚克隆数量和基因组成的计算方法。我们用它来检测急性髓系白血病和乳腺癌样本中的亚克隆,这些亚克隆虽然在疾病发作时就已存在,但从单个原发肿瘤样本中并不明显。通过这样做,我们可以追踪肿瘤进化并确定抵抗治疗的细胞的空间起源。