Sic Heiko, Speletas Matthaios, Cornacchione Vanessa, Seidl Maximillian, Beibel Martin, Linghu Bolan, Yang Fan, Sevdali Eirini, Germenis Anastasios E, Oakeley Edward J, Vangrevelinghe Eric, Sailer Andreas W, Traggiai Elisabetta, Gram Hermann, Eibel Hermann
Novartis Institute for Biomedical Research, Basel, Switzerland.
Department of Immunology and Histocompatibility, Faculty of Medicine, School of Health Sciences, University of Thessaly, Biopolis, Larissa, Greece.
Front Immunol. 2017 Dec 15;8:1824. doi: 10.3389/fimmu.2017.01824. eCollection 2017.
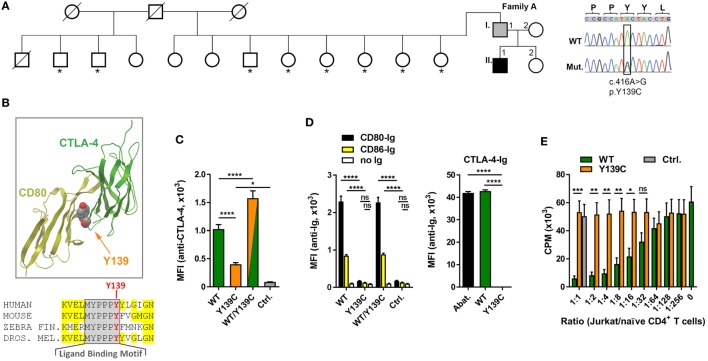
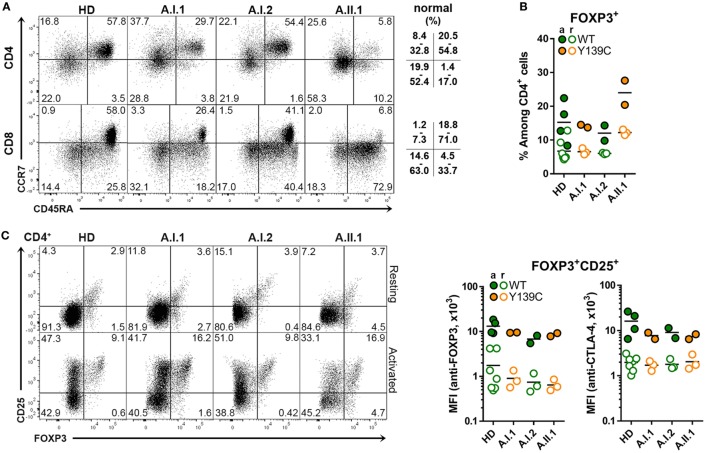
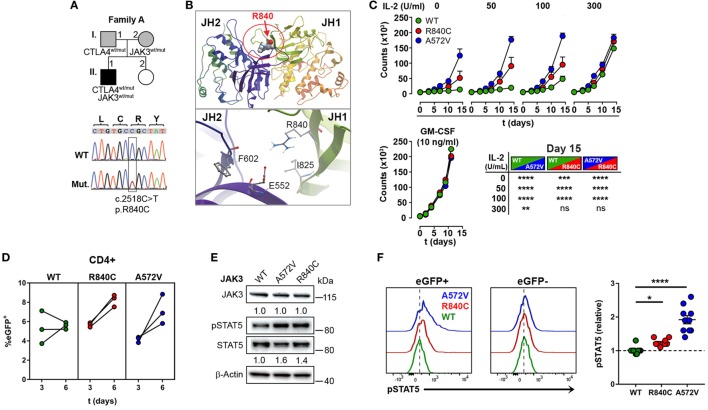
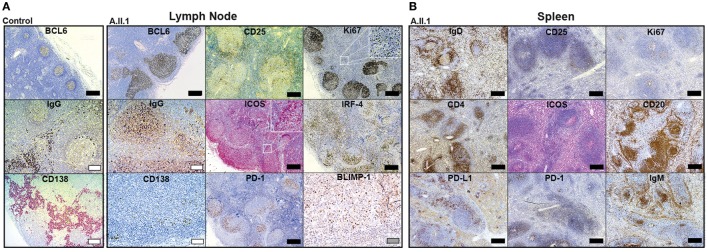
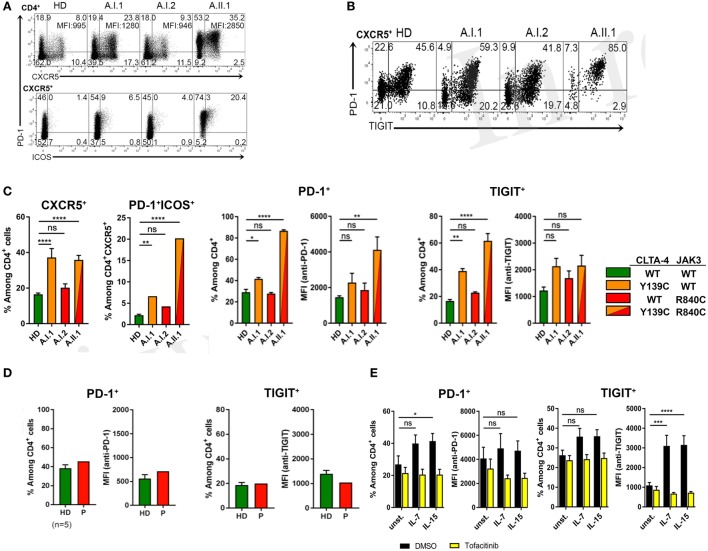
Heterozygous mutations in the cytotoxic T lymphocyte antigen-4 (CTLA-4) are associated with lymphadenopathy, autoimmunity, immune dysregulation, and hypogammaglobulinemia in about 70% of the carriers. So far, the incomplete penetrance of CTLA-4 haploinsufficiency has been attributed to unknown genetic modifiers, epigenetic changes, or environmental effects. We sought to identify potential genetic modifiers in a family with differential clinical penetrance of CTLA-4 haploinsufficiency. Here, we report on a rare heterozygous gain-of-function mutation in Janus kinase-3 (JAK3) (p.R840C), which is associated with the clinical manifestation of CTLA-4 haploinsufficiency in a patient carrying a novel loss-of-function mutation in CTLA-4 (p.Y139C). While the asymptomatic parents carry either the CTLA-4 mutation or the JAK3 variant, their son has inherited both heterozygous mutations and suffers from hypogammaglobulinemia combined with autoimmunity and lymphoid hyperplasia. Although the patient's lymph node and spleen contained many hyperplastic germinal centers with follicular helper T (T) cells and immunoglobulin (Ig) G-positive B cells, plasma cell, and memory B cell development was impaired. CXCR5PD-1TIGIT T cells contributed to a large part of circulating T cells, but they produced only very low amounts of interleukin (IL)-4, IL-10, and IL-21 required for the development of memory B cells and plasma cells. We, therefore, suggest that the combination of the loss-of-function mutation in CTLA-4 with the gain-of-function mutation in JAK3 directs the differentiation of CD4 T cells into dysfunctional T cells supporting the development of lymphadenopathy, hypogammaglobulinemia, and immunodeficiency. Thus, the combination of rare genetic heterozygous variants that remain clinically unnoticed individually may lead to T cell hyperactivity, impaired memory B cell, and plasma cell development resulting finally in combined immunodeficiency.
细胞毒性T淋巴细胞抗原4(CTLA-4)的杂合突变在约70%的携带者中与淋巴结病、自身免疫、免疫失调和低丙种球蛋白血症相关。到目前为止,CTLA-4单倍体不足的不完全外显率一直归因于未知的基因修饰因子、表观遗传变化或环境影响。我们试图在一个CTLA-4单倍体不足具有不同临床外显率的家族中鉴定潜在的基因修饰因子。在此,我们报告了Janus激酶3(JAK3)(p.R840C)中一种罕见的杂合功能获得性突变,该突变与一名携带CTLA-4新功能丧失性突变(p.Y139C)的患者中CTLA-4单倍体不足的临床表现相关。虽然无症状的父母一方携带CTLA-4突变,另一方携带JAK3变体,但他们的儿子同时继承了这两种杂合突变,患有低丙种球蛋白血症并伴有自身免疫和淋巴组织增生。尽管患者的淋巴结和脾脏含有许多增生的生发中心,有滤泡辅助性T(T)细胞和免疫球蛋白(Ig)G阳性B细胞,但浆细胞和记忆B细胞的发育受损。CXCR5⁺PD-1⁺TIGIT⁺ T细胞占循环T细胞的很大一部分,但它们产生的记忆B细胞和浆细胞发育所需的白细胞介素(IL)-4、IL-10和IL-21量极低。因此,我们认为CTLA-4功能丧失性突变与JAK3功能获得性突变的组合将CD4 T细胞分化为功能失调的T细胞,从而导致淋巴结病、低丙种球蛋白血症和免疫缺陷的发展。因此,单独临床上未被注意到的罕见基因杂合变体的组合可能导致T细胞过度活跃、记忆B细胞和浆细胞发育受损,最终导致联合免疫缺陷。