Biomolecular Measurement Division, National Institute of Standards and Technology, 100 Bureau Drive, Gaithersburg, MD, 20899, USA.
Institute for Bioscience and Biotechnology Research, 9600 Gudelsky Drive, Rockville, MD, 20850, USA.
Anal Bioanal Chem. 2018 Mar;410(8):2111-2126. doi: 10.1007/s00216-018-0848-6. Epub 2018 Feb 7.
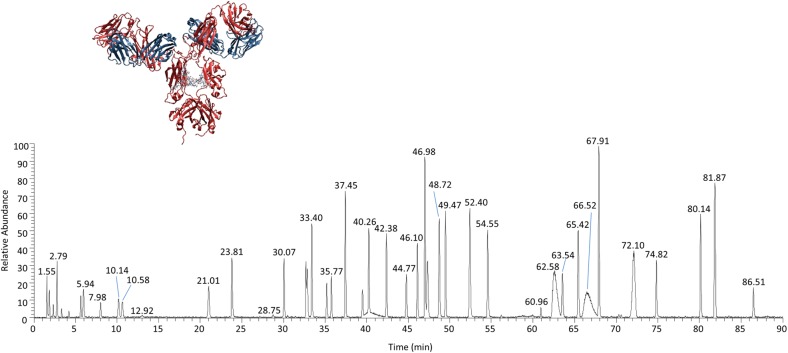
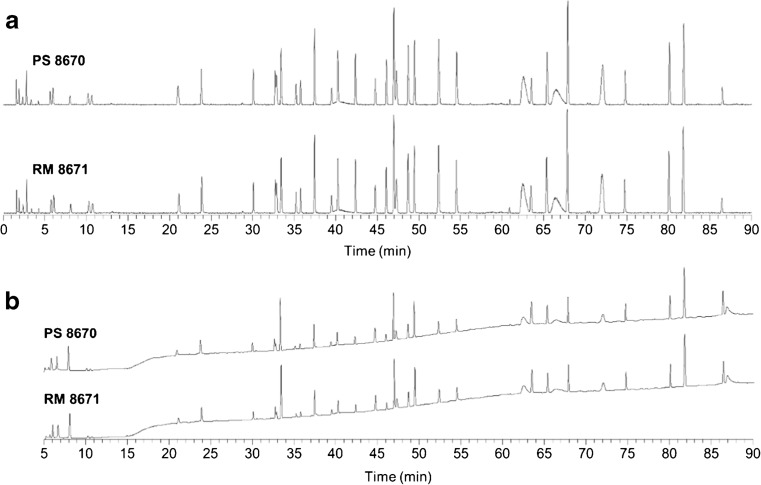
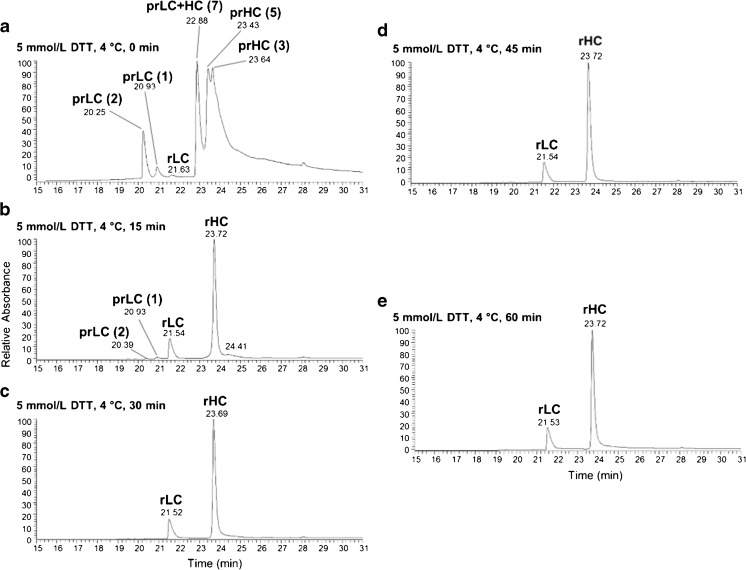
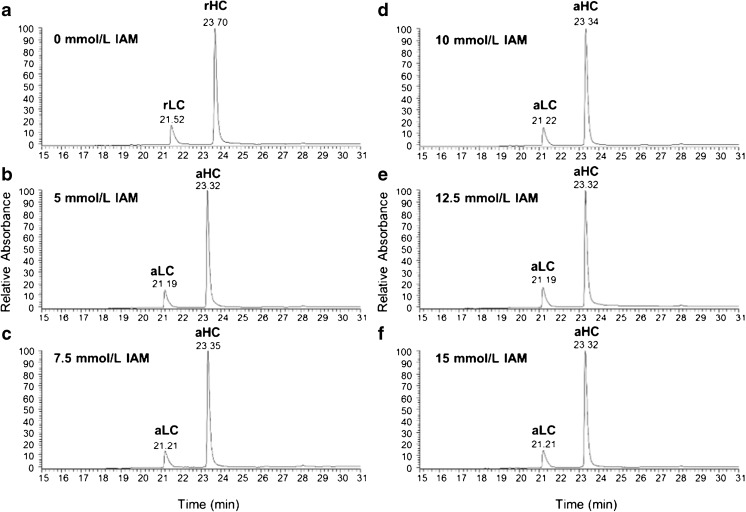
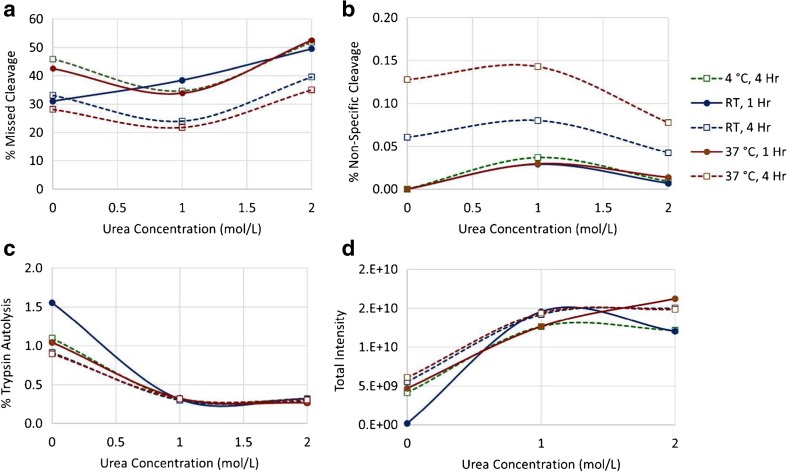
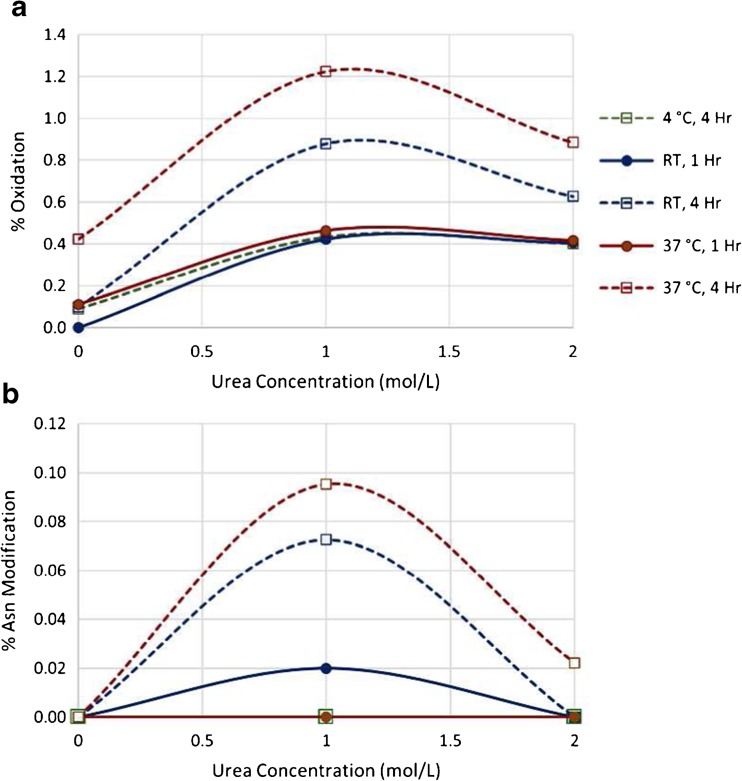
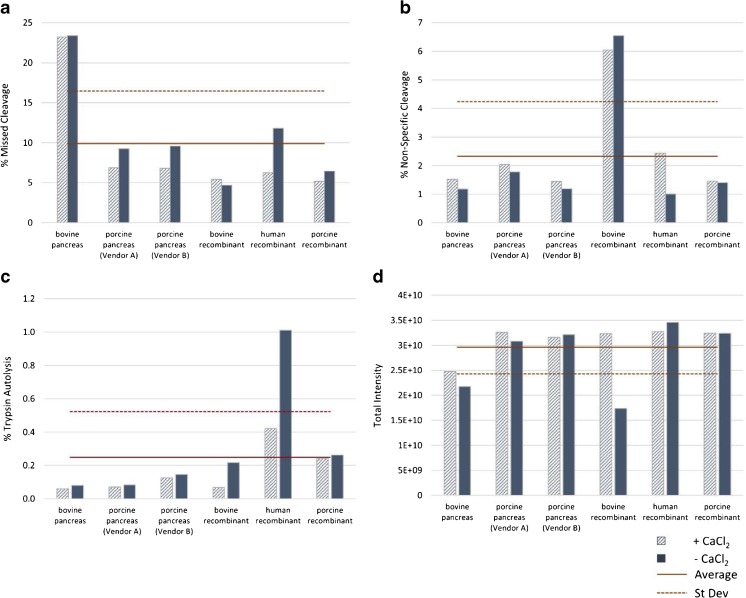
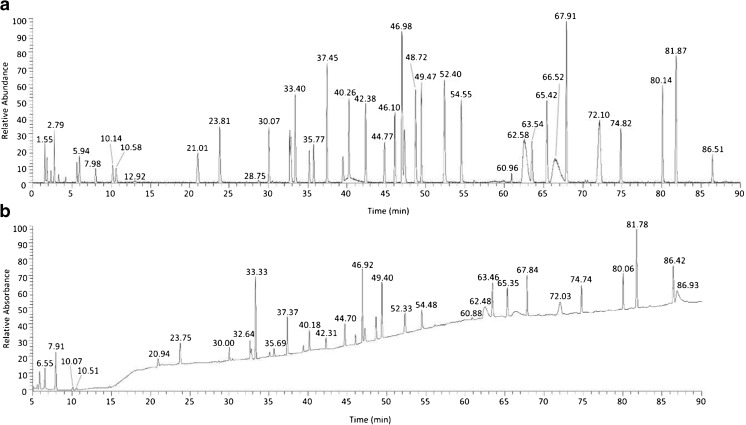
Peptide mapping is a component of the analytical toolbox used within the biopharmaceutical industry to aid in the identity confirmation of a protein therapeutic and to monitor degradative events such as oxidation or deamidation. These methods offer the advantage of providing site-specific information regarding post-translational and chemical modifications that may arise during production, processing or storage. A number of such variations may also be induced by the sample preparation methods themselves which may confound the ability to accurately evaluate the true modification levels. One important focus when developing a peptide mapping method should therefore be the use of sample preparation conditions that will minimize the degree of artificial modifications induced. Unfortunately, the conditions that are amenable to effective reduction, alkylation and digestion are often the same conditions that promote unwanted modifications. Here we describe the optimization of a tryptic digestion protocol used for peptide mapping of the NISTmAb IgG1κ which addresses the challenge of balancing maximum digestion efficiency with minimum artificial modifications. The parameters on which we focused include buffer concentration, digestion time and temperature, as well as the source and type of trypsin (recombinant vs. pancreatic; bovine vs porcine) used. Using the optimized protocol we generated a peptide map of the NISTmAb which allowed us to confirm its identity at the level of primary structure. Graphical abstract Peptide map of the NISTmAb RM 8671 monoclonal antibody. Tryptic digestion was performed using an optimized protocol and followed by LC-UV-MS analysis. The trace represents the total ion chromatogram. Each peak was mapped to peptides identified using mass spectrometry data.
肽图分析是生物制药行业中分析工具包的一个组成部分,用于辅助确认蛋白质治疗药物的身份,并监测氧化或脱酰胺等降解事件。这些方法的优点是提供有关翻译后和化学修饰的特定位置信息,这些修饰可能在生产、加工或储存过程中出现。许多这样的变化也可能是由样品制备方法本身引起的,这可能会干扰准确评估真实修饰水平的能力。因此,开发肽图分析方法的一个重要重点应该是使用能够最大程度减少诱导的人工修饰程度的样品制备条件。不幸的是,易于有效还原、烷基化和消化的条件往往也是促进不需要的修饰的条件。在这里,我们描述了用于 NISTmAb IgG1κ 肽图分析的胰蛋白酶消化方案的优化,该方案解决了在最大消化效率与最小人工修饰之间取得平衡的挑战。我们关注的参数包括缓冲液浓度、消化时间和温度,以及使用的胰蛋白酶(重组 vs 胰脏;牛 vs 猪)的来源和类型。使用优化的方案,我们生成了 NISTmAb 的肽图,这使我们能够在一级结构水平上确认其身份。