Department of Oncology, the First Affiliated Hospital of Xian Jiaotong University, Xi'an, Shaanxi Province, 710061, People's Republic of China.
Department of Biology and Biochemistry, University of Houston, Houston, TX, 77204, USA.
BMC Genomics. 2018 Feb 13;19(1):137. doi: 10.1186/s12864-018-4527-y.
Comprehensive understanding of intratumor heterogeneity requires identification of molecular markers, which are capable of differentiating different subpopulations and which also have clinical significance. One important tool that has been addressing this issue is single cell RNA-Sequencing (scRNASeq) that allows the quantification of expression profiles of transcripts in individual cells in a population of cancer cells. Using the expression profiles from scRNASeq, current studies conduct analysis to group cells into different subpopulations using clustering algorithms. In this study, we explore scRNASeq cancer data from a different perspective. We focus on scRNASeq data originating from cancer cells pertaining to a particular cancer type, where the cell type or the subpopulation to which each cell belongs is known. We investigate if the "cell type" of a cancer cell can be predicted based on the expression profiles of a small set of transcripts.
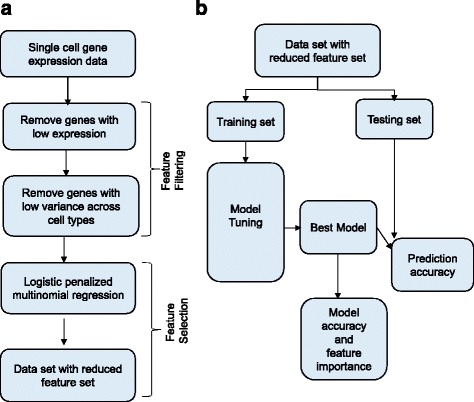
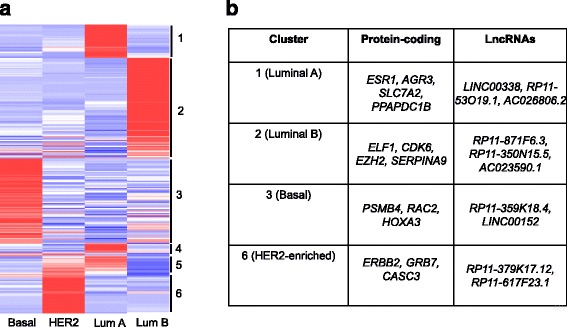
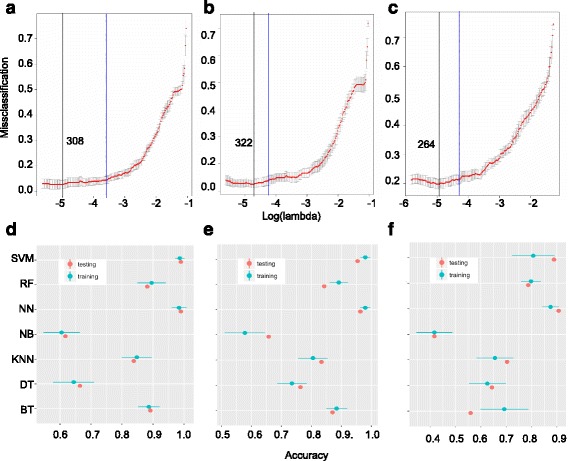
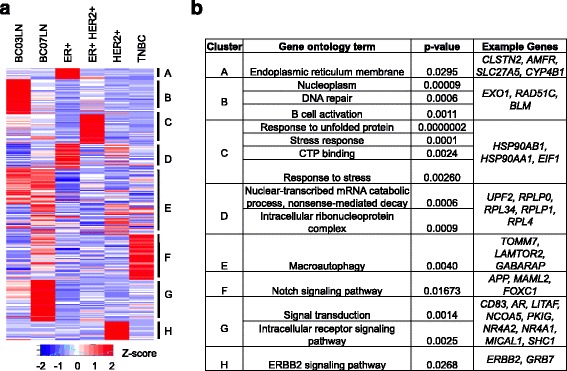
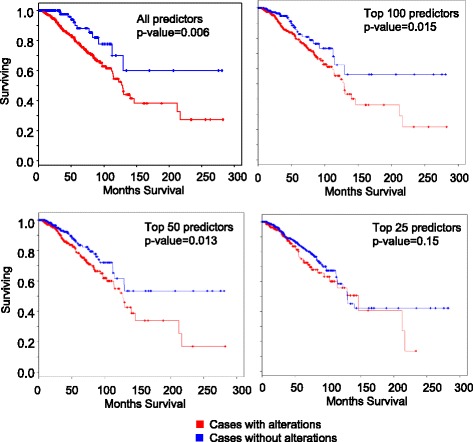
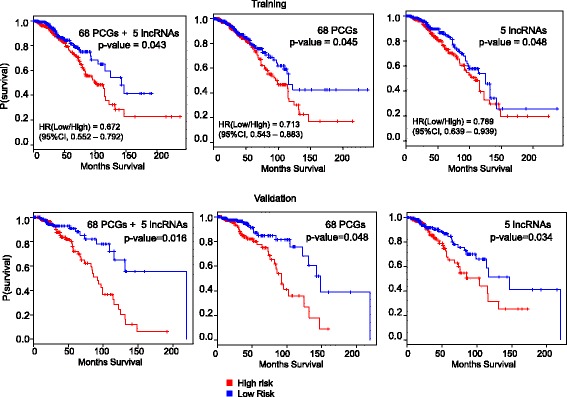
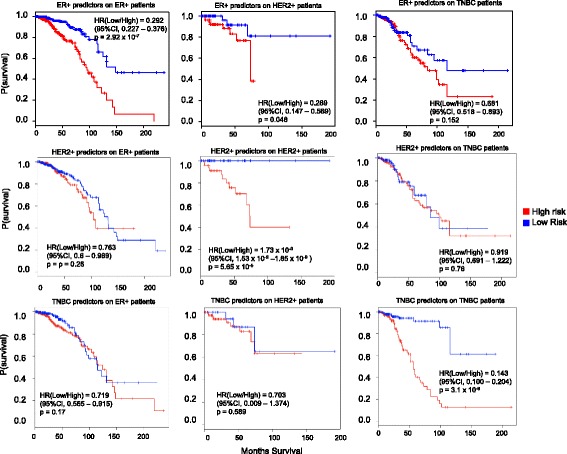
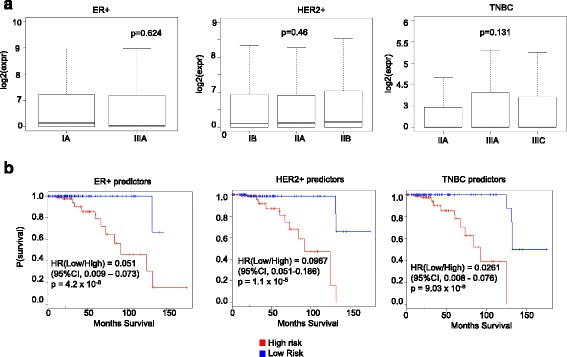
We outline a predictive analytics pipeline to accurately predict 6 breast cancer cell types using single cell gene expression profiles. Instead of building predictive models using the complete human transcripts, the pipeline first eliminates predictors with low expression and low variance. A multinomial penalized logistic regression further reduces the size of the predictors to only 308, out of which 34 are long non-coding RNAs. Tuning of predictive models shows support vector machines and neural networks as the most accurate models achieving close to 98% prediction accuracies. We also find that mixture of protein coding genes and long non-coding RNAs are better predictors compared to when the two sets of transcripts are treated separately. A signature risk score originating from 65 protein coding genes and 5 lncRNA predictors is associated with prognostic survival of TCGA breast cancer patients. This association was maintained when the risk scores were generated using 65 PCGs and 5 lncRNA separately. We further show that predictors restricted to a particular cell type serve as better prognostic markers for the respective patient subtype.
Our results show that in general, the breast cancer cell type predictors are also associated with patient survivability and hence have clinical significance.
全面了解肿瘤内异质性需要识别分子标记物,这些标记物能够区分不同的亚群,并且具有临床意义。解决这个问题的一个重要工具是单细胞 RNA 测序(scRNA-Seq),它允许定量分析癌细胞群体中单个细胞的转录物表达谱。利用 scRNA-Seq 的表达谱,当前的研究使用聚类算法将细胞分为不同的亚群。在这项研究中,我们从不同的角度探索 scRNA-Seq 癌症数据。我们专注于源自特定癌症类型的癌细胞的 scRNA-Seq 数据,其中已知每个细胞所属的细胞类型或亚群。我们研究是否可以根据一小部分转录物的表达谱来预测癌细胞的“细胞类型”。
我们概述了一个预测分析管道,该管道可以使用单细胞基因表达谱准确预测 6 种乳腺癌细胞类型。该管道首先消除表达量低和方差低的预测因子,而不是使用完整的人类转录本构建预测模型。多类惩罚逻辑回归进一步将预测因子的大小缩小到仅 308 个,其中 34 个是长非编码 RNA。预测模型的调整表明,支持向量机和神经网络是最准确的模型,准确率接近 98%。我们还发现,混合蛋白质编码基因和长非编码 RNA 是比单独处理这两组转录本更好的预测因子。源自 65 个蛋白质编码基因和 5 个 lncRNA 预测因子的签名风险评分与 TCGA 乳腺癌患者的预后生存相关。当使用 65 个 PCG 和 5 个 lncRNA 分别生成风险评分时,这种关联仍然存在。我们进一步表明,限制在特定细胞类型的预测因子是各自患者亚型的更好预后标志物。
我们的结果表明,一般来说,乳腺癌细胞类型的预测因子也与患者的存活率相关,因此具有临床意义。