Center for Biological and Engineering Sciences, Sandia National Laboratories, Albuquerque, NM 87185, USA.
Department of Chemical and Biomolecular Engineering, Tulane University, New Orleans, LA 70118, USA.
Top Curr Chem (Cham). 2018 Feb 12;376(2):7. doi: 10.1007/s41061-018-0187-2.
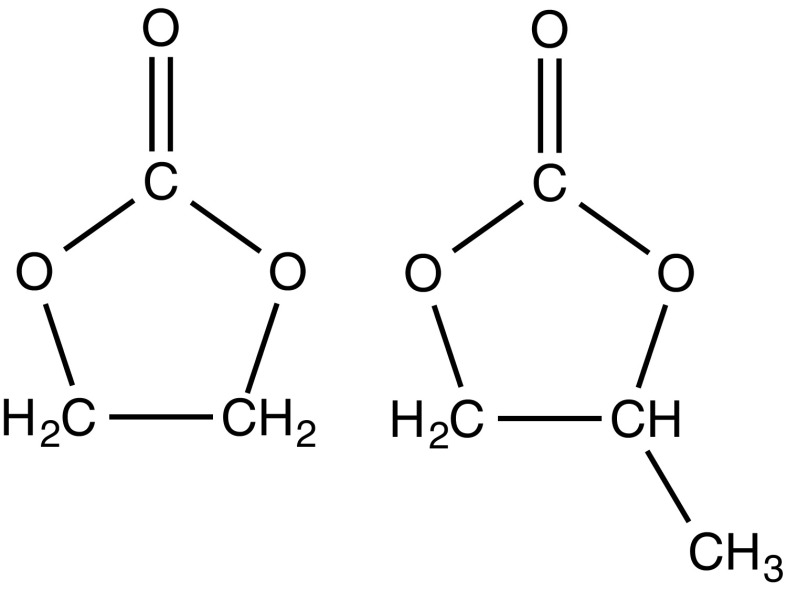
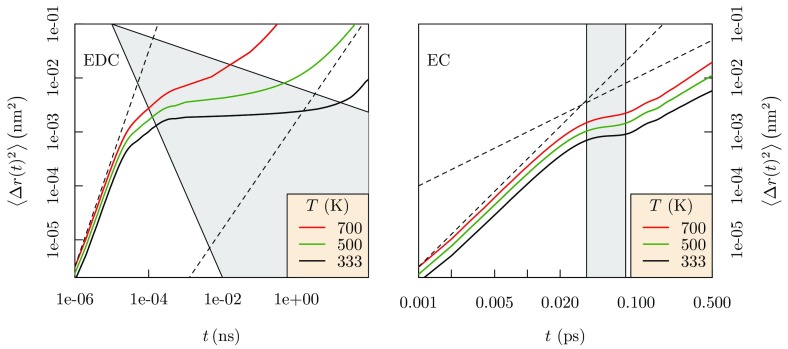
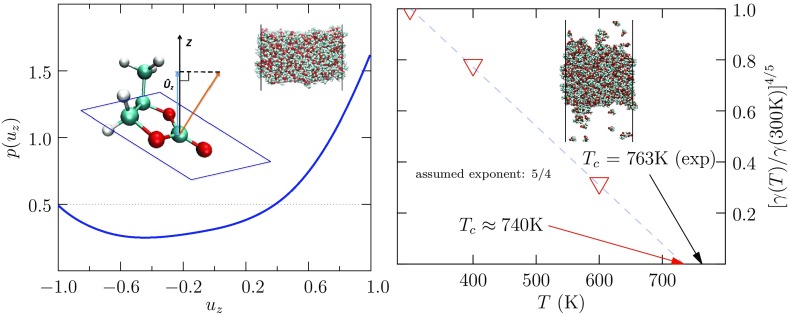
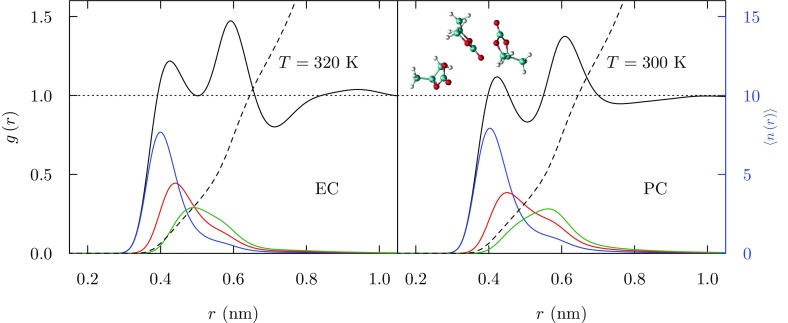
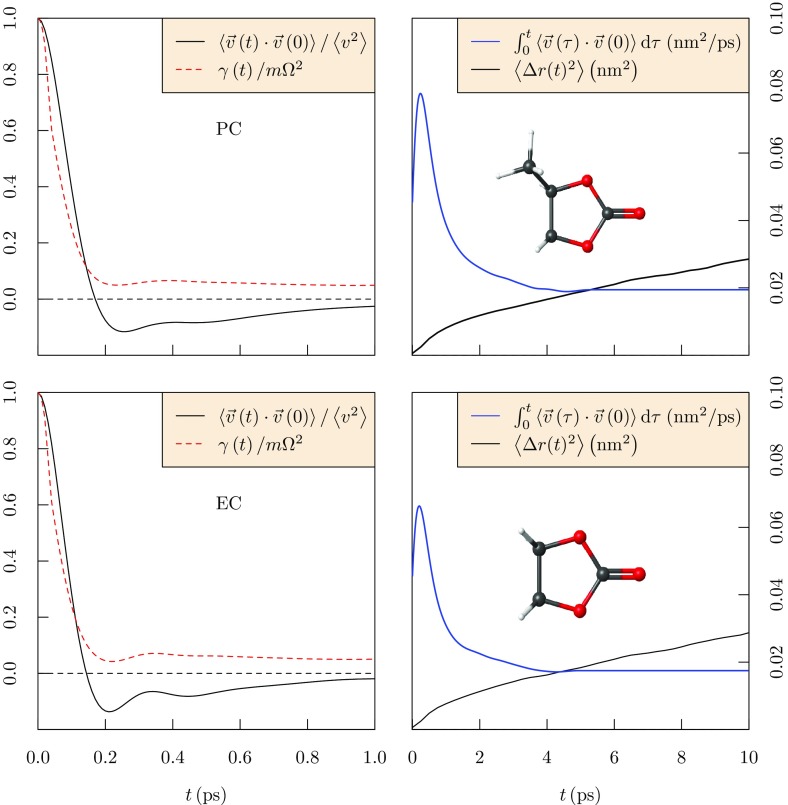
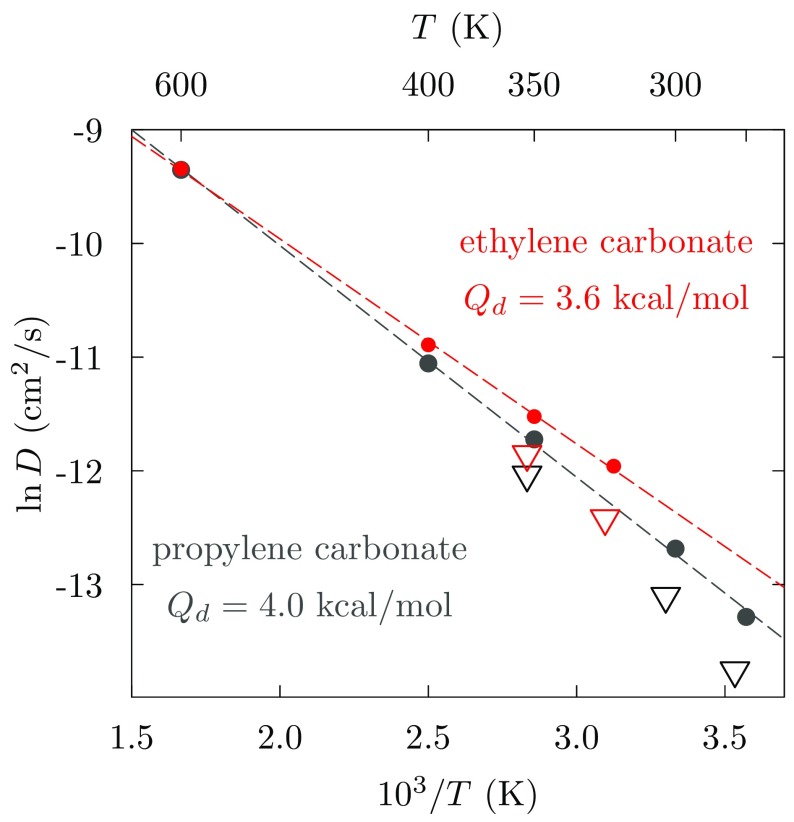
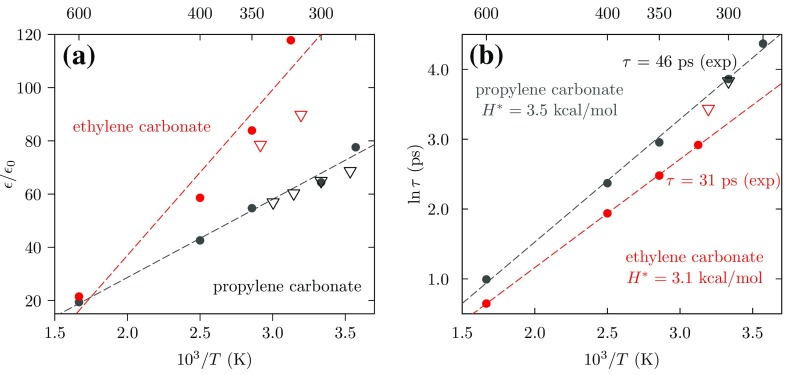
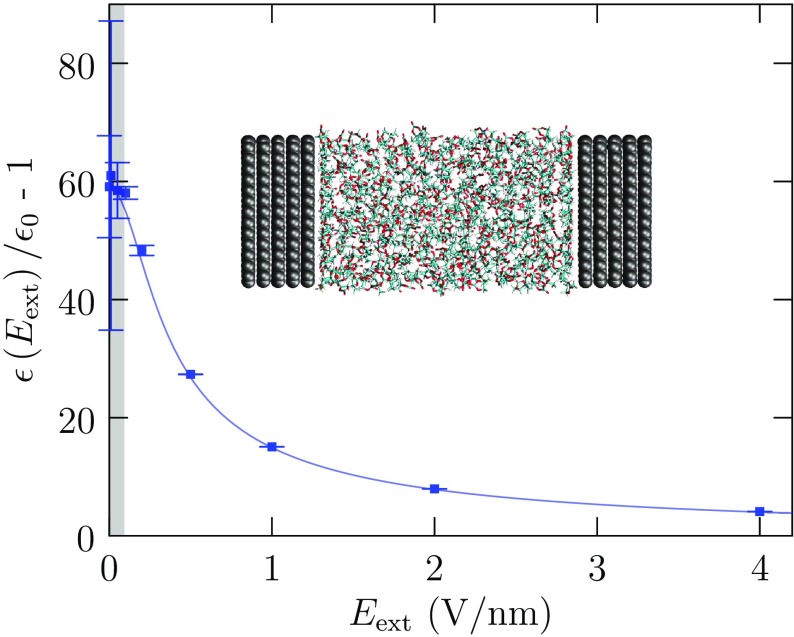
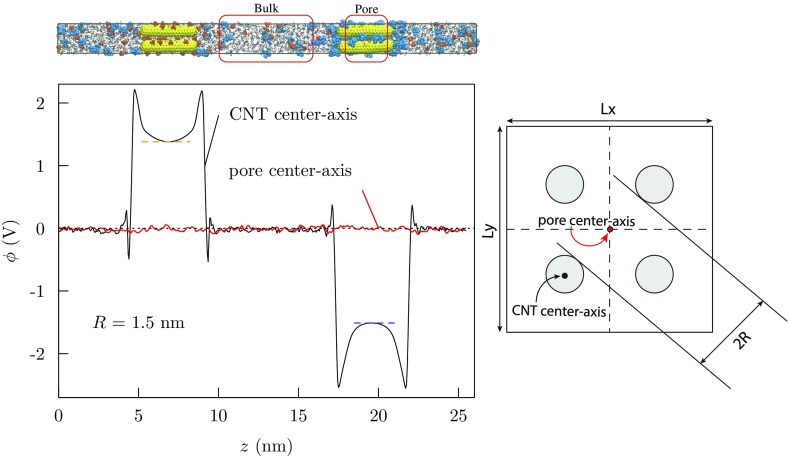
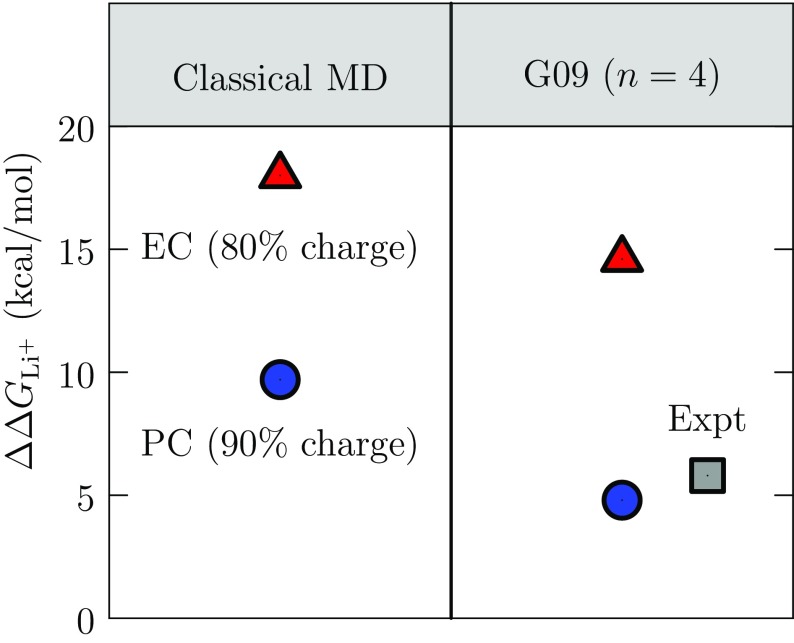
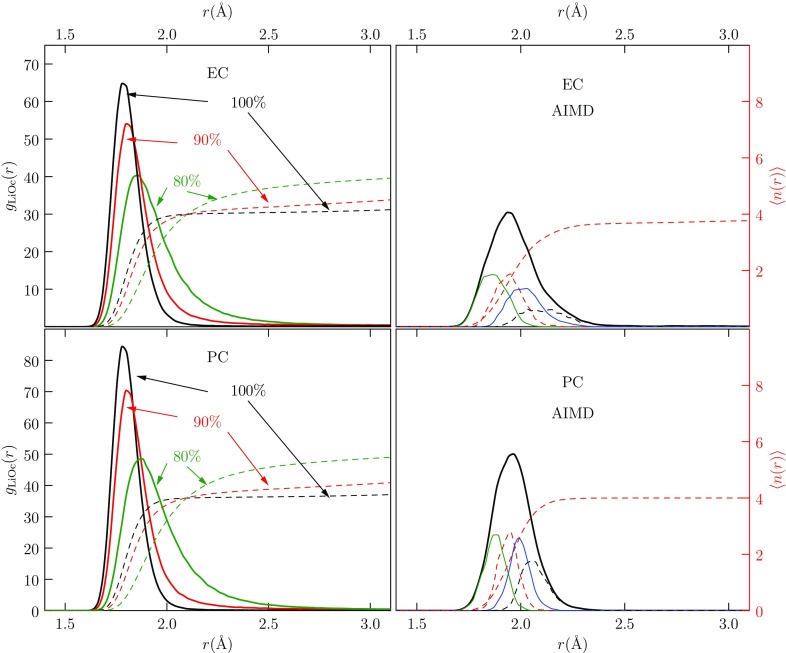
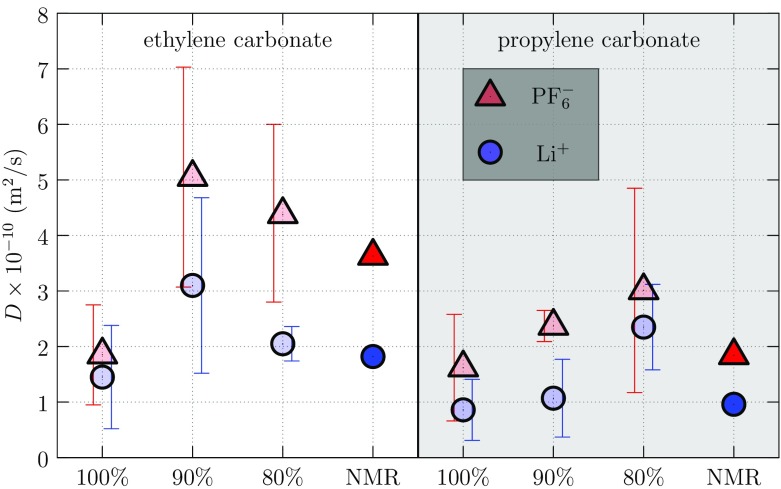
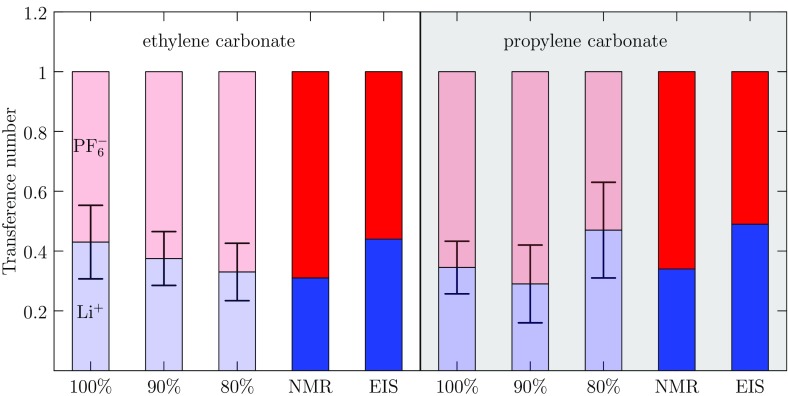
Progress in understanding liquid ethylene carbonate (EC) and propylene carbonate (PC) on the basis of molecular simulation, emphasizing simple models of interatomic forces, is reviewed. Results on the bulk liquids are examined from the perspective of anticipated applications to materials for electrical energy storage devices. Preliminary results on electrochemical double-layer capacitors based on carbon nanotube forests and on model solid-electrolyte interphase (SEI) layers of lithium ion batteries are considered as examples. The basic results discussed suggest that an empirically parameterized, non-polarizable force field can reproduce experimental structural, thermodynamic, and dielectric properties of EC and PC liquids with acceptable accuracy. More sophisticated force fields might include molecular polarizability and Buckingham-model description of inter-atomic overlap repulsions as extensions to Lennard-Jones models of van der Waals interactions. Simple approaches should be similarly successful also for applications to organic molecular ions in EC/PC solutions, but the important case of Li[Formula: see text] deserves special attention because of the particularly strong interactions of that small ion with neighboring solvent molecules. To treat the Li[Formula: see text] ions in liquid EC/PC solutions, we identify interaction models defined by empirically scaled partial charges for ion-solvent interactions. The empirical adjustments use more basic inputs, electronic structure calculations and ab initio molecular dynamics simulations, and also experimental results on Li[Formula: see text] thermodynamics and transport in EC/PC solutions. Application of such models to the mechanism of Li[Formula: see text] transport in glassy SEI models emphasizes the advantage of long time-scale molecular dynamics studies of these non-equilibrium materials.
基于分子模拟,综述了在理解液态碳酸乙烯酯(EC)和碳酸丙烯酯(PC)方面的进展,重点介绍了简单的原子间相互作用力模型。从预期应用于电化学储能器件材料的角度,考察了这些模型在本体液体方面的研究结果。以基于碳纳米管森林的电化学双层电容器和锂离子电池模型固体电解质界面(SEI)层为例,介绍了初步的研究结果。所讨论的基本结果表明,经验参数化的非极化力场可以以可接受的精度再现 EC 和 PC 液体的实验结构、热力学和介电性质。更复杂的力场可能包括分子极化率和原子间重叠排斥的 Buckingham 模型描述,作为范德华相互作用 Lennard-Jones 模型的扩展。对于 EC/PC 溶液中有机分子离子的应用,简单的方法也应该同样成功,但由于该小离子与相邻溶剂分子的相互作用特别强,Li[Formula: see text]的情况值得特别关注。为了在液态 EC/PC 溶液中处理 Li[Formula: see text]离子,我们确定了由离子-溶剂相互作用经验缩放部分电荷定义的相互作用模型。经验调整使用更基本的输入,如电子结构计算和从头算分子动力学模拟,以及 EC/PC 溶液中 Li[Formula: see text]热力学和输运的实验结果。这些模型在玻璃态 SEI 模型中 Li[Formula: see text]输运机制方面的应用强调了对这些非平衡材料进行长时间尺度分子动力学研究的优势。