Pasqualetto Gaia, Brancale Andrea, Young Mark T
School of Pharmacy and Pharmaceutical Sciences, Cardiff University, Cardiff, United Kingdom.
School of Biosciences, Cardiff University, Cardiff, United Kingdom.
Front Pharmacol. 2018 Feb 2;9:58. doi: 10.3389/fphar.2018.00058. eCollection 2018.
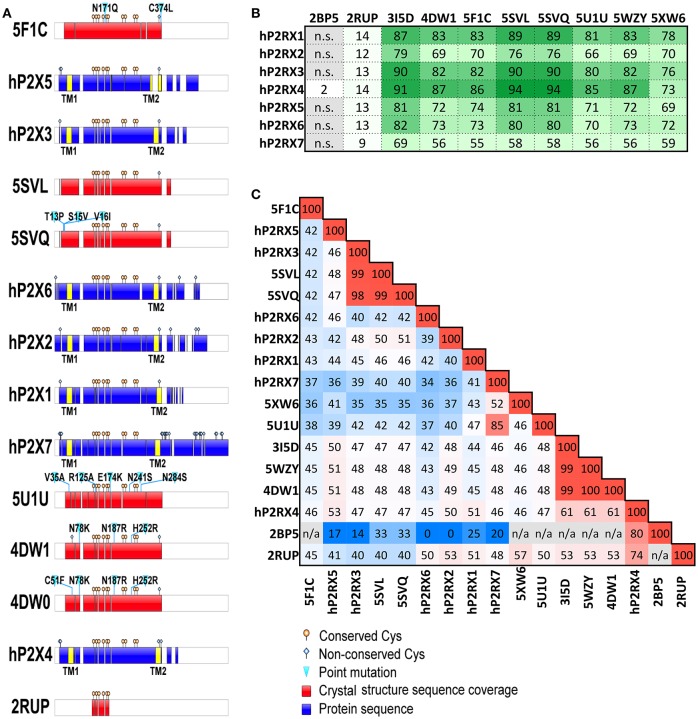
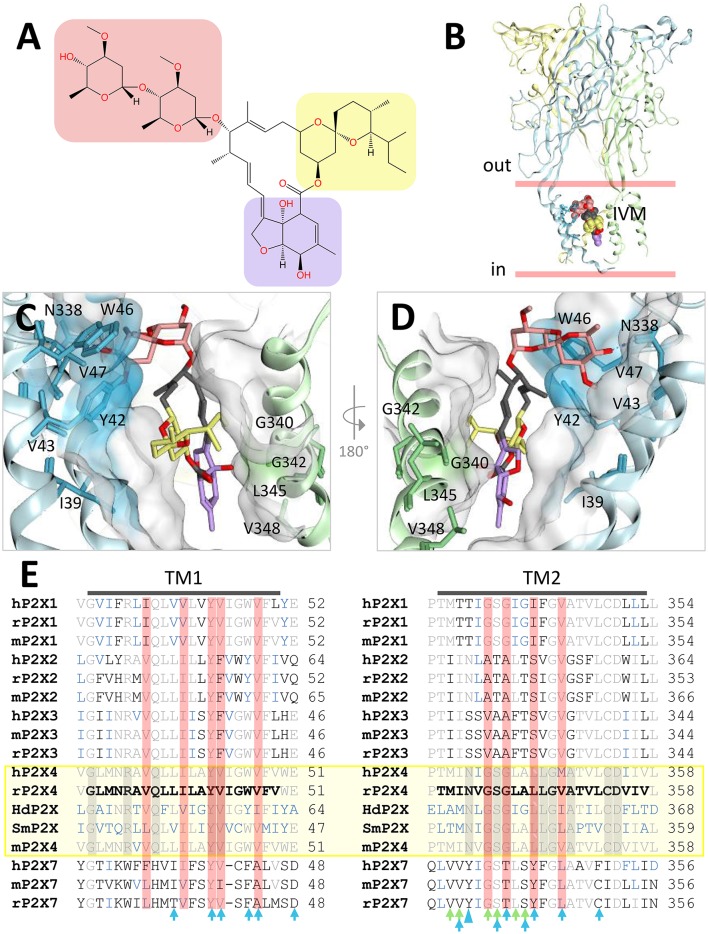
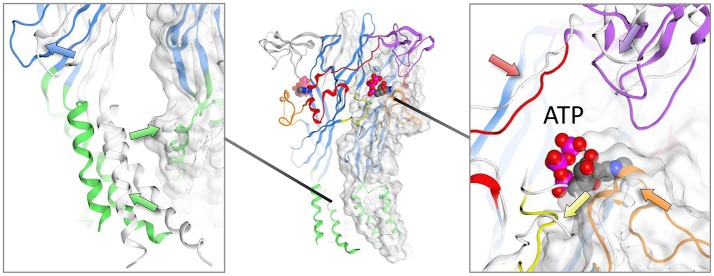
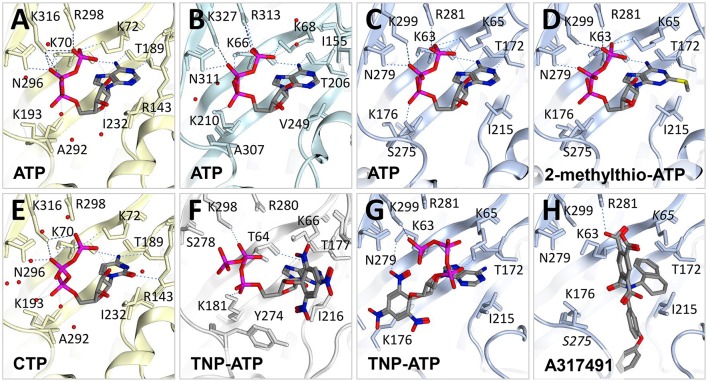
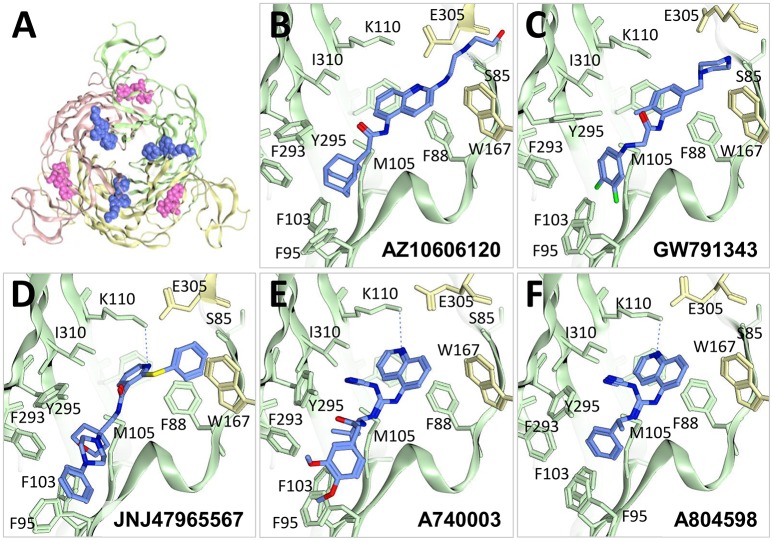
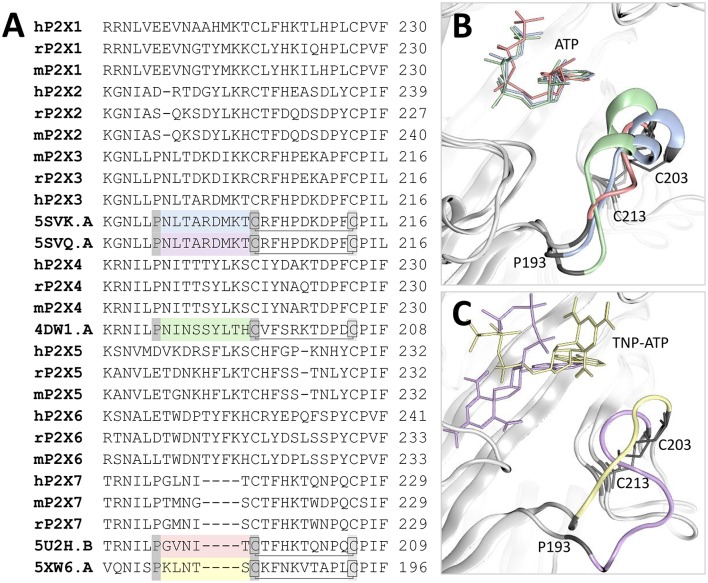
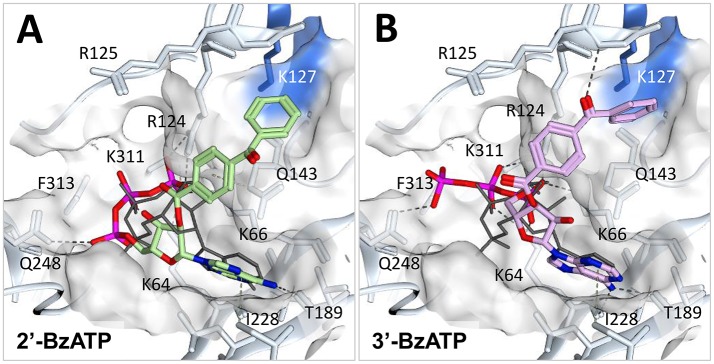
P2X receptors are trimeric eukaryotic ATP-gated cation channels. Extracellular ATP-their physiological ligand-is released as a neurotransmitter and in conditions of cell damage such as inflammation, and substantial evidence implicates P2X receptors in diseases including neuropathic pain, cancer, and arthritis. In 2009, the first P2X crystal structure, P2X4 in the - state, was published, and this was followed in 2012 by the ATP-bound structure. These structures transformed our understanding of the conformational changes induced by ATP binding and the mechanism of ligand specificity, and enabled homology modeling of mammalian P2X receptors for ligand docking and rational design of receptor modulators. P2X receptors are attractive drug targets, and a wide array of potent, subtype-selective modulators (mostly antagonists) have been developed. In 2016, crystal structures of human P2X3 in complex with the competitive antagonists TNP-ATP and A-317491, and P2X7 in complex with a series of allosteric antagonists were published, giving fascinating insights into the mechanism of channel antagonism. In this article we not only summarize current understanding of small-molecule modulator binding at P2X receptors, but also use this information in combination with previously published structure-function data and molecular docking experiments, to hypothesize a role for the dorsal fin loop region in differential ATP potency, and describe novel, testable binding conformations for both the semi-selective synthetic P2X7 agonist 2'-(3')-O-(4-benzoyl)benzoyl ATP (BzATP), and the P2X4-selective positive allosteric modulator ivermectin. We find that the distal benzoyl group of BzATP lies in close proximity to Lys-127, a residue previously implicated in BzATP binding to P2X7, potentially explaining the increased potency of BzATP at rat P2X7 receptors. We also present molecular docking of ivermectin to rat P2X4 receptors, illustrating a plausible binding conformation between the first and second transmembrane domains which not only tallies with previous mutagenesis studies, but would also likely have the effect of stabilizing the open channel structure, consistent with the mode of action of this positive allosteric modulator. From our docking simulations and analysis of sequence homology we propose a series of mutations likely to confer ivermectin sensitivity to human P2X1.
P2X受体是三聚体真核ATP门控阳离子通道。细胞外ATP(其生理配体)作为神经递质释放,并且在诸如炎症等细胞损伤情况下释放,大量证据表明P2X受体与包括神经性疼痛、癌症和关节炎在内的疾病有关。2009年,首个P2X晶体结构,即处于失活状态的P2X4被公布,随后在2012年公布了与ATP结合的结构。这些结构改变了我们对ATP结合诱导的构象变化以及配体特异性机制的理解,并使得能够对哺乳动物P2X受体进行同源建模以用于配体对接和受体调节剂的合理设计。P2X受体是有吸引力的药物靶点,并且已经开发出了各种各样强效的、亚型选择性调节剂(大多为拮抗剂)。2016年,公布了与竞争性拮抗剂TNP - ATP和A - 317491结合的人P2X3以及与一系列变构拮抗剂结合的P2X7的晶体结构,这为通道拮抗机制提供了引人入胜的见解。在本文中,我们不仅总结了目前对P2X受体小分子调节剂结合的理解,还将这些信息与先前发表的结构 - 功能数据和分子对接实验相结合,以推测背鳍环区域在不同ATP效力中的作用,并描述半选择性合成P2X7激动剂2' - (3') - O - (4 - 苯甲酰基)苯甲酰基ATP(BzATP)和P2X4选择性正变构调节剂伊维菌素的新型可测试结合构象。我们发现BzATP的远端苯甲酰基靠近赖氨酸 - 127,该残基先前与BzATP与P2X7的结合有关,这可能解释了BzATP对大鼠P2X7受体效力增加的原因。我们还展示了伊维菌素与大鼠P2X4受体的分子对接,说明了第一和第二跨膜结构域之间一种合理的结合构象,这不仅与先前的诱变研究相符,而且可能具有稳定开放通道结构的作用,这与这种正变构调节剂的作用方式一致。通过我们的对接模拟和序列同源性分析,我们提出了一系列可能使人类P2X1对伊维菌素敏感的突变。