Van Diep Nguyen, Sueyoshi Masuo, Norimine Junzo, Hirai Takuya, Myint Ohnmar, Teh Angeline Ping Ping, Izzati Uda Zahli, Fuke Naoyuki, Yamaguchi Ryoji
Department of Veterinary Medicine, Faculty of Agriculture, University of Miyazaki, Gakuen-kibanadai-nishi-1-1, Miyazaki, 889-2192, Japan.
BMC Vet Res. 2018 Mar 14;14(1):96. doi: 10.1186/s12917-018-1409-0.
Since late 2013, porcine epidemic diarrhea virus (PEDV) has reemerged in Japan and caused severe economic losses to the swine industry. Although PEDV vaccines have been used widely, the disease has swept rapidly across the county, and is commonly observed in PED-vaccinated farms, and has recurred in domestic herds. To better understand PEDVs responsible for the reemerging outbreaks in Japan, full-length spike (S), membrane (M), and nucleocapsid (N) genes of 45 PEDVs collected in Japan during 2013-2016, were sequenced and analyzed.
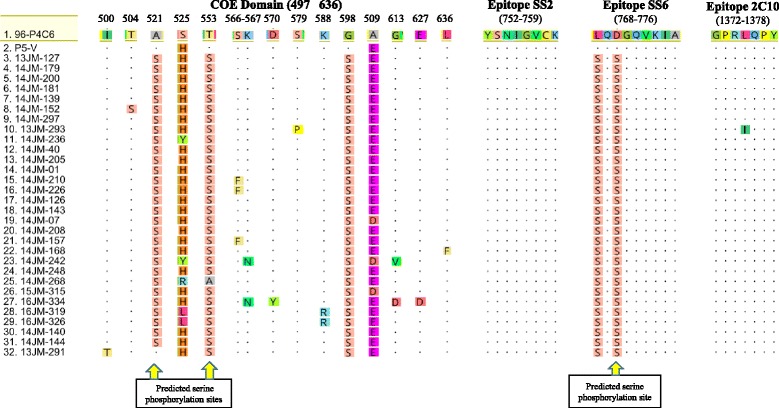
Phylogenetic analysis based on S gene sequences revealed that all the recent field PEDVs were genetically distinct from the classical Japanese strains, and were classified into three genotypes: North American (NA), S INDEL, and Asian non-S INDEL. Our data suggested a possibility that multiple parental PEDV strains were introduced into Japan from abroad at the same time or similar times. The newly identified Japanese strains showed the closest relationship to the US strains. Two sublineages of Japanese strains circulating in Japan were similar to two sublineages identified in the US, suggesting common ancestors for these strains. In comparison with two vaccine strains used in Japan, the field strains had various changes in epitope regions, glycosylation sites, and phosphorylation sites. These substitutions, particularly observed in epitope regions of the S (521, 553, 568, and 570), M (5), and N (123, 252, and 255) proteins, may have affected antigenicity and vaccine efficacy, resulting in an unsuccessful PEDV control. Sequence comparisons between PEDVs collected from primary and secondary outbreaks in three herds revealed that the disease has developed to an endemic stage in which PEDV could persist for nearly two years in the herds or local regions, causing subsequent epidemics.
These results elucidate the genetic characteristics, origin, and molecular epidemiology of PEDVs circulating in Japan, as well as the PEDV strains causing recurrent outbreaks. This study provides a better insight into the PEDVs responsible for recent outbreaks in Japan, and could potentially help to develop measures for controlling and preventing the disease.
自2013年末以来,猪流行性腹泻病毒(PEDV)在日本再度出现,给养猪业造成了严重经济损失。尽管PEDV疫苗已被广泛使用,但该疾病仍迅速席卷全国,在接种过PEDV疫苗的猪场中也普遍存在,且在国内猪群中反复出现。为了更好地了解导致日本再次爆发疫情的PEDV,对2013 - 2016年期间在日本收集的45株PEDV的全长刺突(S)、膜(M)和核衣壳(N)基因进行了测序和分析。
基于S基因序列的系统发育分析表明,所有近期的田间PEDV在基因上与日本经典毒株不同,可分为三种基因型:北美型(NA)、S INDEL型和亚洲非S INDEL型。我们的数据表明,可能有多个亲本PEDV毒株在同一时间或相近时间从国外传入日本。新鉴定出的日本毒株与美国毒株关系最为密切。在日本流行的日本毒株的两个亚系与在美国鉴定出的两个亚系相似,表明这些毒株有共同的祖先。与日本使用的两种疫苗毒株相比,田间毒株在表位区域、糖基化位点和磷酸化位点有多种变化。这些替换,特别是在S(521、553、568和570)、M(5)和N(123、252和255)蛋白的表位区域观察到的替换,可能影响了抗原性和疫苗效力,导致PEDV防控失败。对三个猪群中初次和二次疫情收集的PEDV进行序列比较发现,该疾病已发展到地方流行阶段,其中PEDV可在猪群或局部地区持续近两年,引发后续疫情。
这些结果阐明了在日本流行的PEDV的遗传特征、起源和分子流行病学,以及导致疫情反复爆发的PEDV毒株。本研究为了解导致日本近期疫情的PEDV提供了更好的见解,并可能有助于制定控制和预防该疾病的措施。