Origone Paola, Gotta Fabio, Lamp Merit, Trevisan Lucia, Geroldi Alessandro, Massucco Davide, Grazzini Matteo, Massa Federico, Ticconi Flavia, Bauckneht Matteo, Marchese Roberta, Abbruzzese Giovanni, Bellone Emilia, Mandich Paola
1Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics and Maternal Child Health, University of Genova, c/o DIMI Viale Benedetto XV, 6 - 16132 Genova, Italy.
Medical Genetic Unit, Ospedale Policlinico San Martino, Genova, Italy.
Cerebellum Ataxias. 2018 Mar 14;5:7. doi: 10.1186/s40673-018-0086-x. eCollection 2018.
Spinocerebellar ataxia 17 (SCA17) is one of the most heterogeneous forms of autosomal dominant cerebellar ataxias with a large clinical spectrum which can mimic other movement disorders such as Huntington disease (HD), dystonia and parkinsonism. SCA17 is caused by an expansion of CAG/CAA repeat in the Tata binding protein () gene. Normal alleles contain 25 to 40 CAG/CAA repeats, alleles with 50 or greater CAG/CAA repeats are pathological with full penetrance. Alleles with 43 to 49 CAG/CAA repeats were also reported and their penetrance is estimated between 50 and 80%. Recently few symptomatic individuals having 41 and 42 repeats were reported but it is still unclear whether CAG/CAA repeats of 41 or 42 are low penetrance disease-causing alleles. Thus, phenotypic variability like the disease course in subject with SCA17 locus restricted expansions remains to be fully understood.
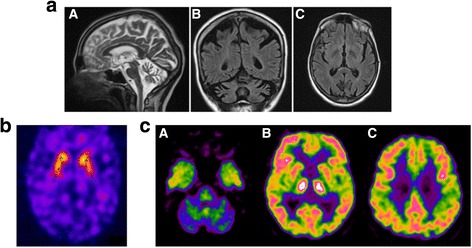
The patients was a 63-year-old woman who, at 54 years, showed personality changes and increased frequency of falls. At 55 years of age neuropsychological tests showed executive attention and visuospatial deficit. At the age of 59 the patient developed dysarthria and a progressive cognitive deficit. The neurological examination showed moderate gait ataxia, dysdiadochokinesia and dysmetria, dysphagia, dysarthria and abnormal saccadic pursuit, severe axial asynergy during postural changes, choreiform dyskinesias. Molecular analysis of the gene demonstrated an allele with 41 repeat suggesting that 41 CAG/CCG repeats could be an allele associated with the full clinical spectrum of SCA17.
The described case with the other similar cases described in the literature suggests that 41 CAG/CAA trinucleotides should be considered as critical threshold in SCA17. We suggest that SCA17 diagnosis should be suspected in patients presenting with movement disorders associated with other neurodegenerative signs and symptoms.
脊髓小脑共济失调17型(SCA17)是常染色体显性小脑共济失调中最具异质性的类型之一,临床谱广泛,可模仿其他运动障碍,如亨廷顿病(HD)、肌张力障碍和帕金森症。SCA17由Tata结合蛋白(TBP)基因中的CAG/CAA重复序列扩增引起。正常等位基因含有25至40个CAG/CAA重复序列,具有50个或更多CAG/CAA重复序列的等位基因具有完全外显率,呈病理性。也有报道含有43至49个CAG/CAA重复序列的等位基因,其外显率估计在50%至80%之间。最近,有少数含有41和42个重复序列的有症状个体被报道,但41或42个CAG/CAA重复序列是否为低外显率致病等位基因仍不清楚。因此,SCA17基因座限制性扩增患者的表型变异性,如病程,仍有待充分了解。
患者为一名63岁女性,54岁时出现性格改变和跌倒频率增加。55岁时神经心理学测试显示执行注意力和视觉空间缺陷。59岁时患者出现构音障碍和进行性认知缺陷。神经系统检查显示中度步态共济失调、轮替运动障碍和辨距不良、吞咽困难、构音障碍和异常扫视追踪、姿势改变时严重的轴性协同不能、舞蹈样运动障碍。TBP基因的分子分析显示一个含有41个重复序列的等位基因,提示41个CAG/CCG重复序列可能是与SCA17完整临床谱相关的等位基因。
本文所述病例及文献中描述的其他类似病例表明,41个CAG/CAA三核苷酸应被视为SCA17的临界阈值。我们建议,对于出现与其他神经退行性体征和症状相关的运动障碍的患者,应怀疑SCA17诊断。