Niu Zhiyv, Pontifex Carly Sabine, Berini Sarah, Hamilton Leslie E, Naddaf Elie, Wieben Eric, Aleff Ross A, Martens Kristina, Gruber Angela, Engel Andrew G, Pfeffer Gerald, Milone Margherita
Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN, United States.
Department of Clinical Genomics, Mayo Clinic, Rochester, MN, United States.
Front Neurol. 2018 Mar 19;9:147. doi: 10.3389/fneur.2018.00147. eCollection 2018.
The aim of this study is to identify the molecular defect of three unrelated individuals with late-onset predominant distal myopathy; to describe the spectrum of phenotype resulting from the contributing role of two variants in genes located on two different chromosomes; and to highlight the underappreciated complex forms of genetic myopathies.
Clinical and laboratory data of three unrelated probands with predominantly distal weakness manifesting in the sixth-seventh decade of life, and available affected and unaffected family members were reviewed. Next-generation sequencing panel, whole exome sequencing, and targeted analyses of family members were performed to elucidate the genetic etiology of the myopathy.
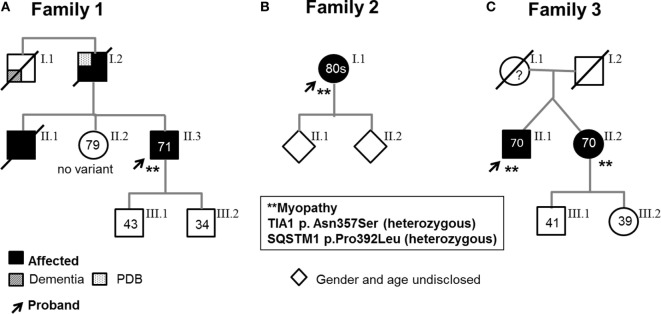
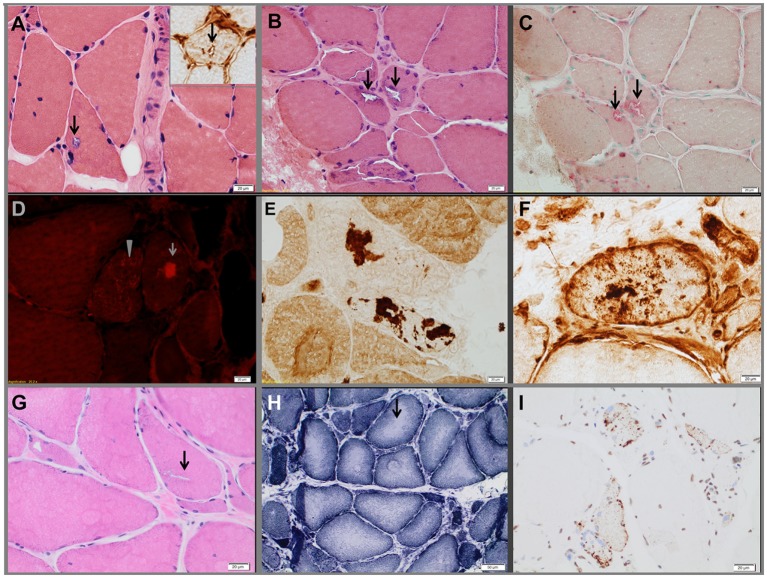
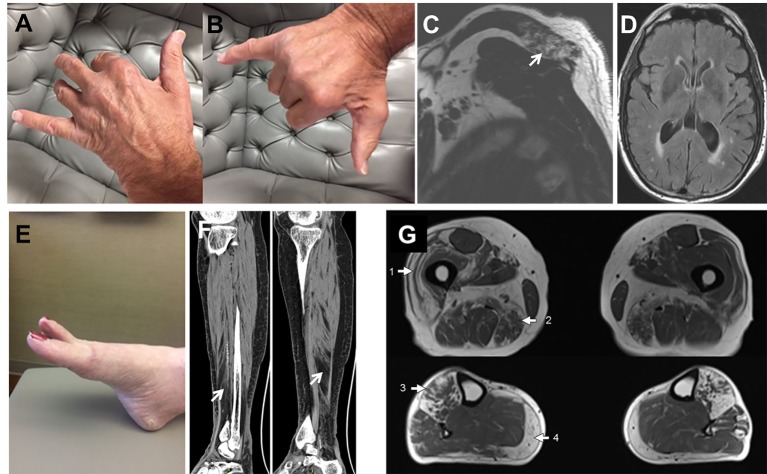
Genetic analyses detected two contributing variants located on different chromosomes in three unrelated probands: a heterozygous pathogenic mutation in (c.1175C>T, p.Pro392Leu) and a heterozygous variant in (c.1070A>G, p.Asn357Ser). The affected fraternal twin of one proband also carries both variants, while the unaffected family members harbor one or none. Two unrelated probands (family 1, II.3, and family 3, II.1) have a distal myopathy with rimmed vacuoles that manifested with index extensor weakness; the other proband (family 2, I.1) has myofibrillar myopathy manifesting with hypercapnic respiratory insufficiency and distal weakness.
The findings indicate that all the affected individuals have a myopathy associated with both variants in and , respectively, suggesting that the two variants determine the phenotype and likely functionally interact. We speculate that the variant is a modifier of the mutation. We identify the combination of and variants as a novel genetic defect associated with myofibrillar myopathy and suggest to consider sequencing both genes in the molecular investigation of myopathy with rimmed vacuoles and myofibrillar myopathy although additional studies are needed to investigate the digenic nature of the disease.
本研究旨在确定三名散发性迟发性以远端为主的肌病患者的分子缺陷;描述位于两条不同染色体上的基因中的两个变异相互作用所导致的表型谱;并强调未得到充分认识的复杂形式的遗传性肌病。
回顾了三名散发性先证者的临床和实验室数据,这些患者主要表现为远端肌无力,症状出现在60 - 70岁,还纳入了其健在的患病和未患病家庭成员。采用二代测序panel、全外显子组测序及对家庭成员进行靶向分析,以阐明该肌病的遗传病因。
基因分析在三名散发性先证者中检测到位于不同染色体上的两个相互作用的变异:一个杂合致病性突变(c.1175C>T,p.Pro392Leu)和一个杂合变异(c.1070A>G,p.Asn357Ser)。一名先证者的患病异卵双胞胎也携带这两个变异,而未患病家庭成员携带其中一个或均不携带。两名散发性先证者(家系1的II.3和家系3的II.1)患有伴有镶边空泡的远端肌病,表现为示指伸肌无力;另一名先证者(家系2的I.1)患有肌原纤维肌病,表现为高碳酸血症性呼吸功能不全和远端肌无力。
研究结果表明,所有患病个体均患有一种分别与和两个变异相关的肌病,提示这两个变异决定了表型,且可能在功能上相互作用。我们推测变异是突变的修饰因子。我们确定和变异的组合是一种与肌原纤维肌病相关的新型遗传缺陷,并建议在对伴有镶边空泡的肌病和肌原纤维肌病进行分子研究时考虑对这两个基因进行测序,尽管还需要更多研究来探究该疾病的双基因性质。