Institute of Neuropathology, RWTH Aachen University, Aachen, Germany.
Department of Neurology, Kassel School of Medicine, Klinikum Kassel, Kassel, Germany.
Brain Pathol. 2020 Sep;30(5):877-896. doi: 10.1111/bpa.12864. Epub 2020 Jun 15.
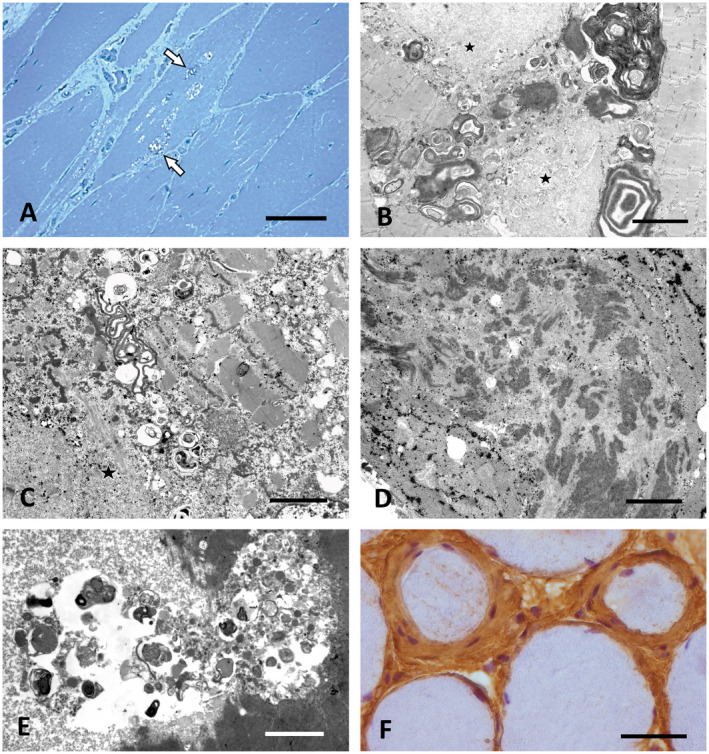
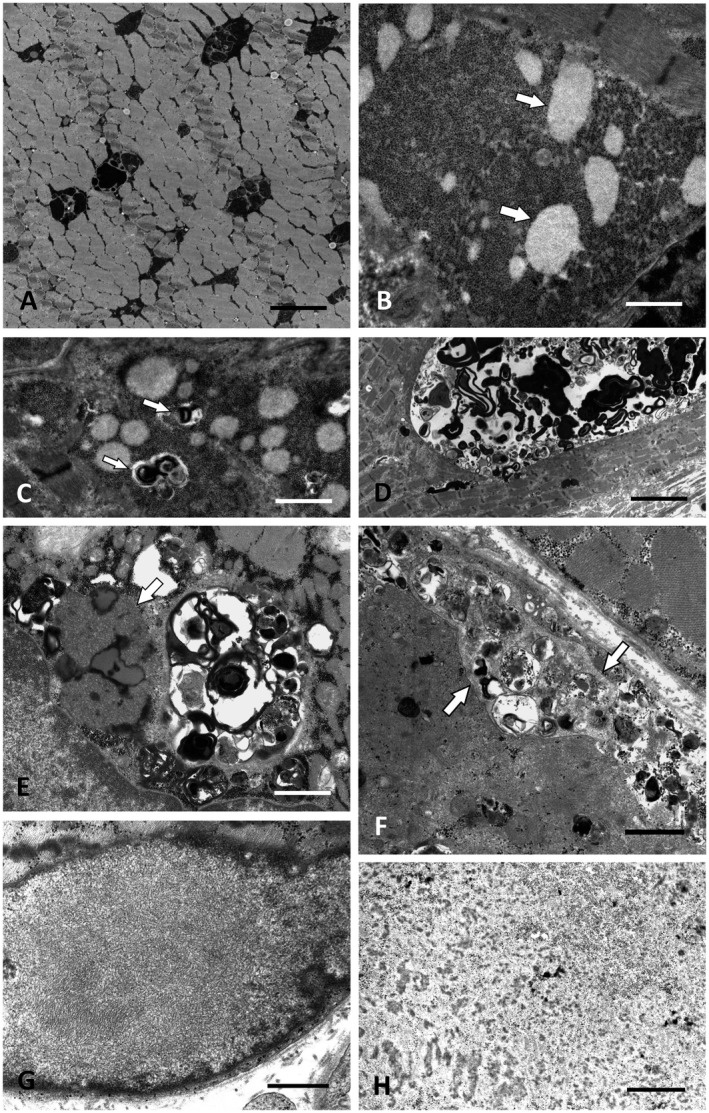
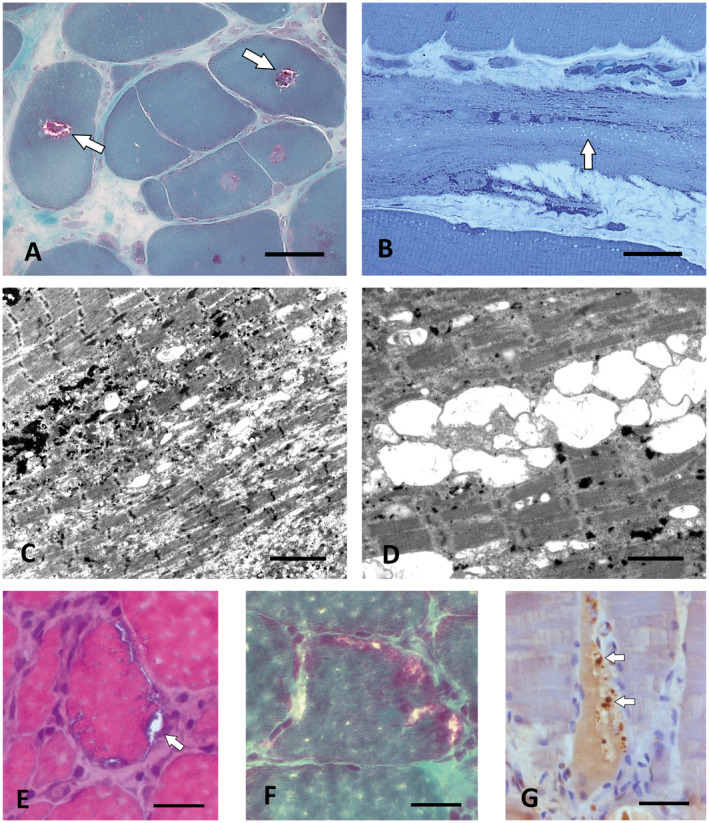
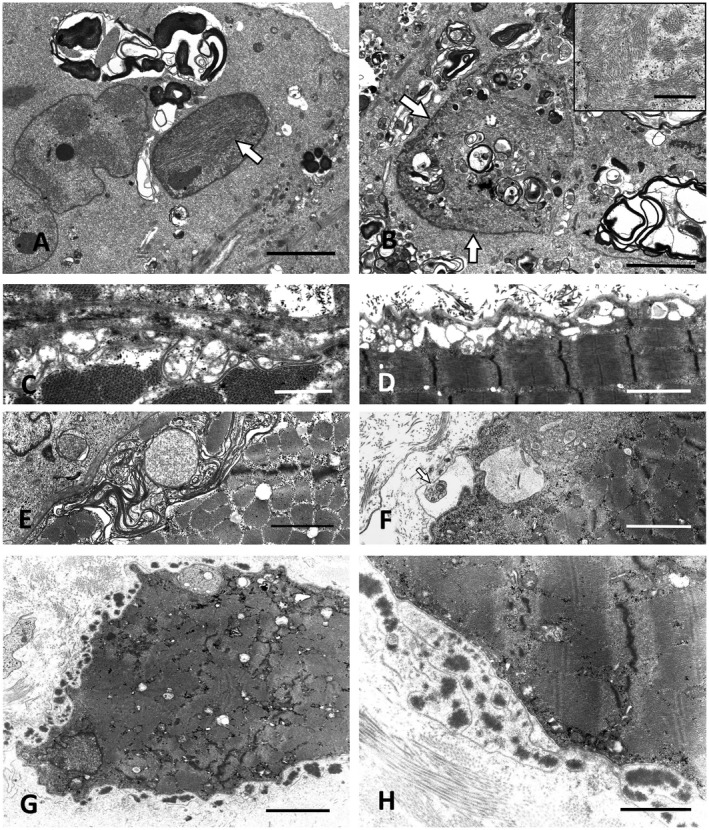
Altered autophagy accompanied by abnormal autophagic (rimmed) vacuoles detectable by light and electron microscopy is a common denominator of many familial and sporadic non-inflammatory muscle diseases. Even in the era of next generation sequencing (NGS), late-onset vacuolar myopathies remain a diagnostic challenge. We identified 32 adult vacuolar myopathy patients from 30 unrelated families, studied their clinical, histopathological and ultrastructural characteristics and performed genetic testing in index patients and relatives using Sanger sequencing and NGS including whole exome sequencing (WES). We established a molecular genetic diagnosis in 17 patients. Pathogenic mutations were found in genes typically linked to vacuolar myopathy (GNE, LDB3/ZASP, MYOT, DES and GAA), but also in genes not regularly associated with severely altered autophagy (FKRP, DYSF, CAV3, COL6A2, GYG1 and TRIM32) and in the digenic facioscapulohumeral muscular dystrophy 2. Characteristic histopathological features including distinct patterns of myofibrillar disarray and evidence of exocytosis proved to be helpful to distinguish causes of vacuolar myopathies. Biopsy validated the pathogenicity of the novel mutations p.(Phe55*) and p.(Arg216*) in GYG1 and of the p.(Leu156Pro) TRIM32 mutation combined with compound heterozygous deletion of exon 2 of TRIM32 and expanded the phenotype of Ala93Thr-caveolinopathy and of limb-girdle muscular dystrophy 2i caused by FKRP mutation. In 15 patients no causal variants were detected by Sanger sequencing and NGS panel analysis. In 12 of these cases, WES was performed, but did not yield any definite mutation or likely candidate gene. In one of these patients with a family history of muscle weakness, the vacuolar myopathy was eventually linked to chloroquine therapy. Our study illustrates the wide phenotypic and genotypic heterogeneity of vacuolar myopathies and validates the role of histopathology in assessing the pathogenicity of novel mutations detected by NGS. In a sizable portion of vacuolar myopathy cases, it remains to be shown whether the cause is hereditary or degenerative.
自噬改变伴可见于光镜和电镜的异常自噬(边缘)空泡是许多家族性和散发性非炎症性肌肉疾病的共同特征。即使在下一代测序(NGS)时代,迟发性空泡性肌病仍然是一个诊断挑战。我们从 30 个无关家庭中鉴定了 32 名成年空泡性肌病患者,研究了他们的临床、组织病理学和超微结构特征,并在索引患者和亲属中使用 Sanger 测序和包括全外显子组测序(WES)的 NGS 进行了基因检测。我们在 17 名患者中建立了分子遗传学诊断。在通常与空泡性肌病相关的基因(GNE、LDB3/ZASP、MYOT、DES 和 GAA)中发现了致病性突变,但也在与严重自噬改变不相关的基因(FKRP、DYSF、CAV3、COL6A2、GYG1 和 TRIM32)以及二基因面肩肱型肌营养不良症 2 中发现了致病性突变。特征性组织病理学特征包括肌原纤维排列紊乱的不同模式和胞吐作用的证据,被证明有助于区分空泡性肌病的病因。活检证实了 GYG1 中的新型突变 p.(Phe55*)和 p.(Arg216*)以及 TRIM32 突变的致病性,该突变与 TRIM32 外显子 2 的复合杂合缺失和扩展的 Ala93Thr-caveolinopathy 表型以及 FKRP 突变引起的肢带型肌营养不良症 2i 相关。在 15 名患者中,通过 Sanger 测序和 NGS 面板分析均未检测到致病变异。在其中 12 例患者中进行了 WES,但未发现任何明确的突变或可能的候选基因。在 1 例有肌肉无力家族史的患者中,空泡性肌病最终与氯喹治疗有关。我们的研究说明了空泡性肌病的广泛表型和基因型异质性,并验证了组织病理学在评估 NGS 检测到的新型突变的致病性中的作用。在相当一部分空泡性肌病病例中,仍有待证明病因是遗传性的还是退行性的。