Department of Chemistry and Chemical Biology, Northeastern University, Boston, Massachusetts.
College of Computer and Information Science, Northeastern University, Boston, Massachusetts.
Protein Sci. 2018 Jun;27(6):1125-1135. doi: 10.1002/pro.3416. Epub 2018 Apr 27.
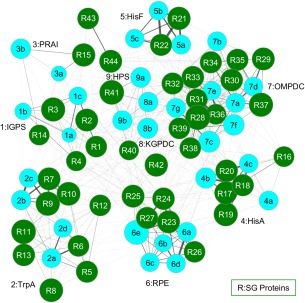
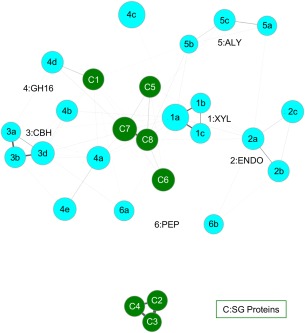
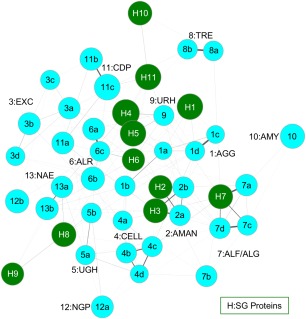
As a result of high-throughput protein structure initiatives, over 14,400 protein structures have been solved by Structural Genomics (SG) centers and participating research groups. While the totality of SG data represents a tremendous contribution to genomics and structural biology, reliable functional information for these proteins is generally lacking. Better functional predictions for SG proteins will add substantial value to the structural information already obtained. Our method described herein, Graph Representation of Active Sites for Prediction of Function (GRASP-Func), predicts quickly and accurately the biochemical function of proteins by representing residues at the predicted local active site as graphs rather than in Cartesian coordinates. We compare the GRASP-Func method to our previously reported method, Structurally Aligned Local Sites of Activity (SALSA), using the Ribulose Phosphate Binding Barrel (RPBB), 6-Hairpin Glycosidase (6-HG), and Concanavalin A-like Lectins/Glucanase (CAL/G) superfamilies as test cases. In each of the superfamilies, SALSA and the much faster method GRASP-Func yield similar correct classification of previously characterized proteins, providing a validated benchmark for the new method. In addition, we analyzed SG proteins using our SALSA and GRASP-Func methods to predict function. Forty-one SG proteins in the RPBB superfamily, nine SG proteins in the 6-HG superfamily, and one SG protein in the CAL/G superfamily were successfully classified into one of the functional families in their respective superfamily by both methods. This improved, faster, validated computational method can yield more reliable predictions of function that can be used for a wide variety of applications by the community.
由于高通量蛋白质结构计划,超过 14400 个蛋白质结构已被结构基因组学(SG)中心和参与的研究小组解决。虽然 SG 数据的总和代表了对基因组学和结构生物学的巨大贡献,但这些蛋白质通常缺乏可靠的功能信息。对 SG 蛋白进行更好的功能预测将为已经获得的结构信息增添巨大的价值。我们在此描述的方法,即活性位点的图形表示用于功能预测(GRASP-Func),通过将预测的局部活性位点处的残基表示为图形而不是笛卡尔坐标,快速准确地预测蛋白质的生化功能。我们将 GRASP-Func 方法与我们之前报道的方法,即结构对齐的局部活性位点(SALSA)进行比较,使用核酮糖磷酸结合桶(RPBB)、6-发夹糖苷酶(6-HG)和伴刀豆球蛋白 A 样凝集素/葡聚糖酶(CAL/G)超家族作为测试案例。在每个超家族中,SALSA 和更快的方法 GRASP-Func 对以前表征的蛋白质进行了相似的正确分类,为新方法提供了有效的基准。此外,我们使用 SALSA 和 GRASP-Func 方法对 SG 蛋白进行了功能预测分析。RPBB 超家族中的 41 个 SG 蛋白、6-HG 超家族中的 9 个 SG 蛋白和 CAL/G 超家族中的 1 个 SG 蛋白都被两种方法成功地分类到各自超家族的功能家族之一。这种改进的、更快的、经过验证的计算方法可以产生更可靠的功能预测,社区可以将其用于各种应用。