Research Group for Ancient Genomics and Evolution, Department of Molecular Biology, Max Planck Institute for Developmental Biology, Tuebingen, Germany.
The Sainsbury Laboratory, Norwich, UK.
BMC Bioinformatics. 2018 Apr 4;19(1):122. doi: 10.1186/s12859-018-2128-z.
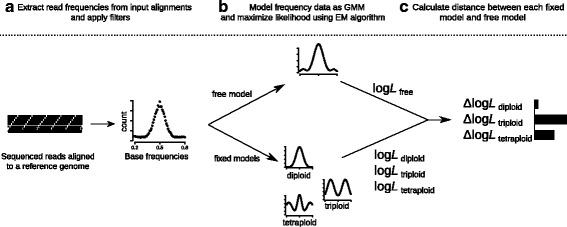
Intraspecific variation in ploidy occurs in a wide range of species including pathogenic and nonpathogenic eukaryotes such as yeasts and oomycetes. Ploidy can be inferred indirectly - without measuring DNA content - from experiments using next-generation sequencing (NGS). We present nQuire, a statistical framework that distinguishes between diploids, triploids and tetraploids using NGS. The command-line tool models the distribution of base frequencies at variable sites using a Gaussian Mixture Model, and uses maximum likelihood to select the most plausible ploidy model. nQuire handles large genomes at high coverage efficiently and uses standard input file formats.
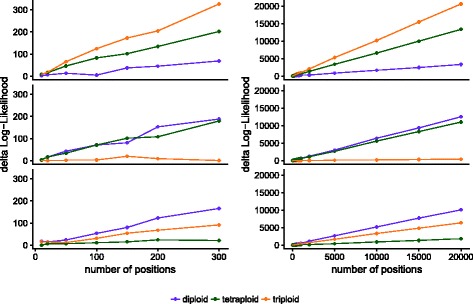
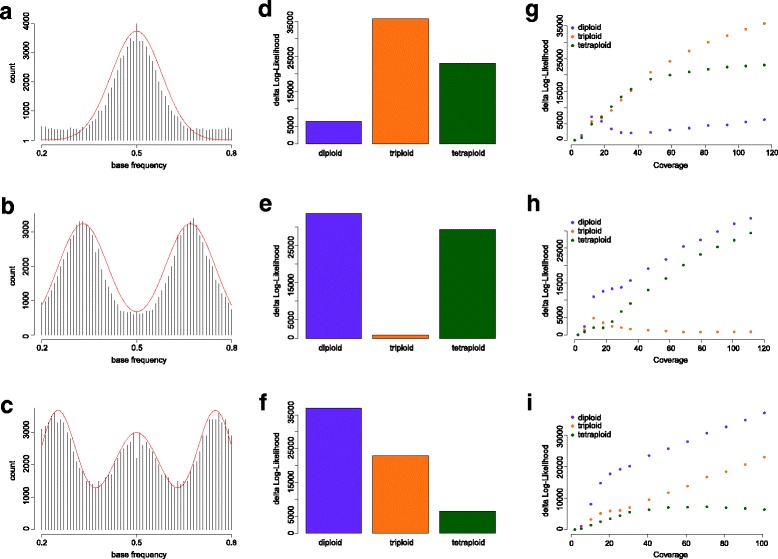
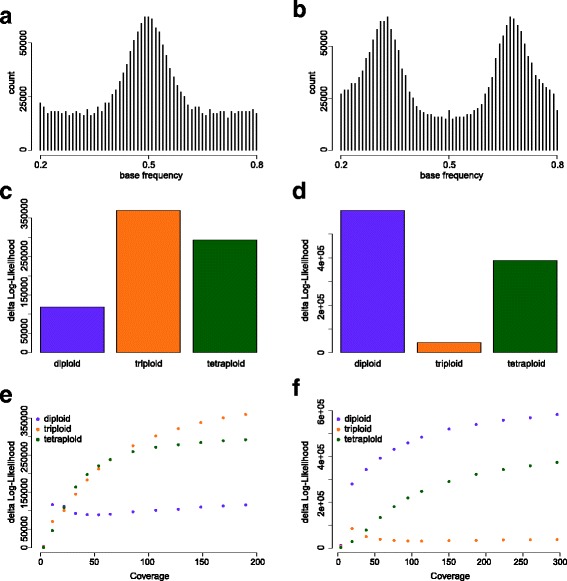
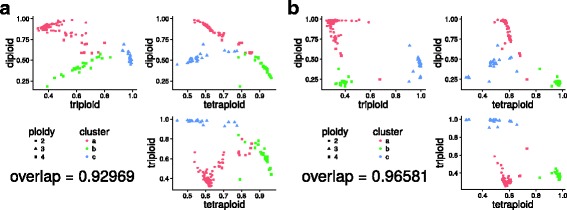
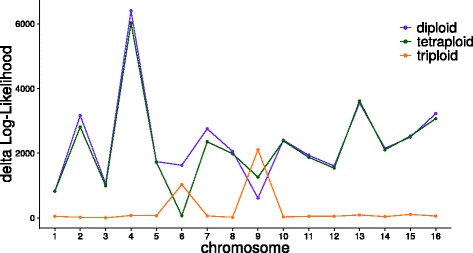
We demonstrate the utility of nQuire analyzing individual samples of the pathogenic oomycete Phytophthora infestans and the Baker's yeast Saccharomyces cerevisiae. Using these organisms we show the dependence between reliability of the ploidy assignment and sequencing depth. Additionally, we employ normalized maximized log- likelihoods generated by nQuire to ascertain ploidy level in a population of samples with ploidy heterogeneity. Using these normalized values we cluster samples in three dimensions using multivariate Gaussian mixtures. The cluster assignments retrieved from a S. cerevisiae population recovered the true ploidy level in over 96% of samples. Finally, we show that nQuire can be used regionally to identify chromosomal aneuploidies.
nQuire provides a statistical framework to study organisms with intraspecific variation in ploidy. nQuire is likely to be useful in epidemiological studies of pathogens, artificial selection experiments, and for historical or ancient samples where intact nuclei are not preserved. It is implemented as a stand-alone Linux command line tool in the C programming language and is available at https://github.com/clwgg/nQuire under the MIT license.
多倍体在广泛的物种中存在种内变异,包括致病性和非致病性真核生物,如酵母和卵菌。多倍体可以通过使用下一代测序(NGS)的实验间接推断出来——而无需测量 DNA 含量。我们提出了 nQuire,这是一种使用 NGS 区分二倍体、三倍体和四倍体的统计框架。该命令行工具使用高斯混合模型对变异位点的碱基频率分布进行建模,并使用最大似然法选择最合理的倍性模型。nQuire 可以高效地处理高覆盖率的大型基因组,并使用标准输入文件格式。
我们通过分析致病性卵菌 Phytophthora infestans 和贝克酵母 Saccharomyces cerevisiae 的单个样本来证明 nQuire 的实用性。使用这些生物体,我们展示了倍性分配的可靠性与测序深度之间的依赖性。此外,我们还使用 nQuire 生成的标准化最大化对数似然来确定具有倍性异质性的样本群体中的倍性水平。使用这些标准化值,我们使用多维高斯混合在三个维度上对样本进行聚类。从 S. cerevisiae 群体中检索到的样本聚类分配在超过 96%的样本中恢复了真实的倍性水平。最后,我们表明 nQuire 可以用于区域内识别染色体非整倍性。
nQuire 提供了一个统计框架来研究具有种内多倍体变异的生物体。nQuire 可能在病原体的流行病学研究、人工选择实验中有用,并且对于没有完整核的历史或古代样本也有用。它是用 C 编程语言实现的独立的 Linux 命令行工具,并在 MIT 许可证下可在 https://github.com/clwgg/nQuire 上获得。