Tewari Vishal V, Mehta Ritu, Sreedhar C M, Tewari Kunal, Mohammad Akbar, Gupta Neerja, Gulati Sheffali, Kabra Madhulika
Departments of Pediatrics, Army Hospital (Referral & Research), New Delhi, 110010, India.
Department of Pathology, All India Institute of Medical Sciences, New Delhi, India.
BMC Pediatr. 2018 Apr 4;18(1):126. doi: 10.1186/s12887-018-1108-9.
4H syndrome is a congenital hypomyelinating leukodystrophy characterized by hypodontia, hypomyelination and hypogonadotropic hypogonadism belonging to the Pol III-related leukodystrophies which arise due to mutations in the POLR3A or POLR3B gene. The clinical presentation is of neurodevelopmental delay or regression with ataxia, dystonia, nystagmus, delayed deciduous dentition and abnormal order of eruption of teeth. MRI brain shows a characteristic hypomyelination pattern. Several mutations have been described in the implicated genes but there are no reports on mutations seen in patients from India.
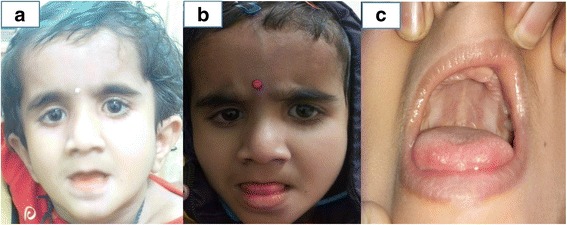
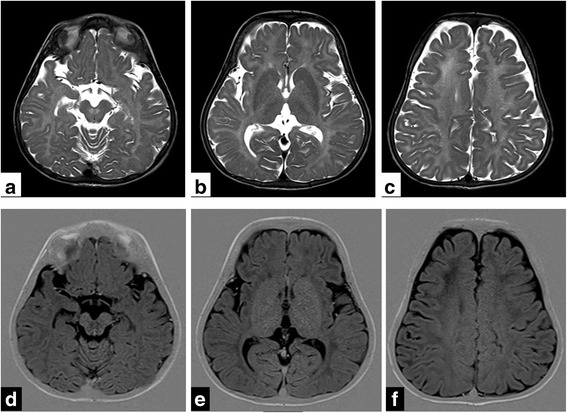
We report a 1½ year old girl, only child of a non-consanguinous couple who presented with delayed developmental milestones and delayed dentition. On physical examination she had downward slanting palpebral fissures, low set ears, smooth philtrum, hypodontia, prominent body hair and clitoromegaly. There was prominent horizontal nystagmus, hypertonia of both upper and lower limbs, exaggerated deep tendon jerks and flexor planter response. She had not attained complete head control and required support to sit. She showed absent waves on brainstem evoked response audiometry and her fundus examination showed bilateral optic atrophy with prolongation of P100 latencies on visual evoked potentials. MRI Brain showed hyperintensity of entire white matter with involvement of the internal and external capsule, frontal deep white matter and corpus callosum. Her karyotype was 46 XX and her endocrinal profile was unremarkable. Clinical exome sequencing identified an unreported mutation in the POLR3A gene. The same mutation was identified by Sanger sequencing in heterozygous state in both parents. The child is being managed with physiotherapy and developmental therapy. She has been provided with hearing aids and started on speech therapy. Parents were provided anticipatory guidance and genetic counselling about autosomal recessive nature of inheritance, risk of recurrence and need for follow-up.
4H syndrome is a rare congenital hypomyelinating leukodystrophy inherited as an autosomal recessive disorder due to mutations in the POLR3A and POLR3B gene. Delay or regression of milestones, abnormalities in dentition and endocrinal perturbations are its hallmark. A novel mutation in the POLR3A gene resulting in amino acid substitution of arginine for glutamine at codon 808 (p.R808Q) was detected in exon 18 in our case.
4H综合征是一种先天性髓鞘形成不良性脑白质营养不良,其特征为牙发育不全、髓鞘形成不良和低促性腺激素性性腺功能减退,属于与聚合酶III相关的脑白质营养不良,由POLR3A或POLR3B基因突变引起。临床表现为神经发育延迟或倒退,伴有共济失调、肌张力障碍、眼球震颤、乳牙萌出延迟和牙齿萌出顺序异常。脑部MRI显示特征性的髓鞘形成不良模式。已在相关基因中描述了几种突变,但尚无来自印度患者的突变报道。
我们报告一名1岁半女童,是非近亲结婚夫妇的独生女,表现为发育里程碑延迟和出牙延迟。体格检查发现她有睑裂向下倾斜、耳朵低位、人中平滑、牙发育不全、体毛浓密和阴蒂肥大。有明显的水平眼球震颤,上下肢张力亢进,深腱反射亢进和跖屈反射。她尚未完全控制头部,需要支撑才能坐立。脑干听觉诱发电位显示无波形,眼底检查显示双侧视神经萎缩,视觉诱发电位P100潜伏期延长。脑部MRI显示整个白质高信号,累及内囊、外囊、额叶深部白质和胼胝体。她的核型为46 XX,内分泌检查无异常。临床外显子测序在POLR3A基因中发现了一个未报道的突变。通过桑格测序在父母双方中均检测到该突变以杂合状态存在。该患儿正在接受物理治疗和发育治疗。已为她提供了助听器并开始进行言语治疗。向父母提供了关于常染色体隐性遗传性质、复发风险和随访必要性的预期指导和遗传咨询。
4H综合征是一种罕见的先天性髓鞘形成不良性脑白质营养不良,由于POLR3A和POLR3B基因突变而作为常染色体隐性疾病遗传。发育里程碑延迟或倒退、牙列异常和内分泌紊乱是其特征。在我们的病例中,在外显子18中检测到POLR3A基因的一个新突变,导致密码子808处精氨酸被谷氨酰胺取代(p.R808Q)。