Department for Medical Genome Sciences, Medical Genome Center, National Center for Geriatrics and Gerontology, Aichi, Japan.
Department of Medical Science Mathematics, Medical Research Institute, Tokyo Medical and Dental University (TMDU), Tokyo, Japan.
Sci Rep. 2018 Apr 4;8(1):5608. doi: 10.1038/s41598-018-23978-z.
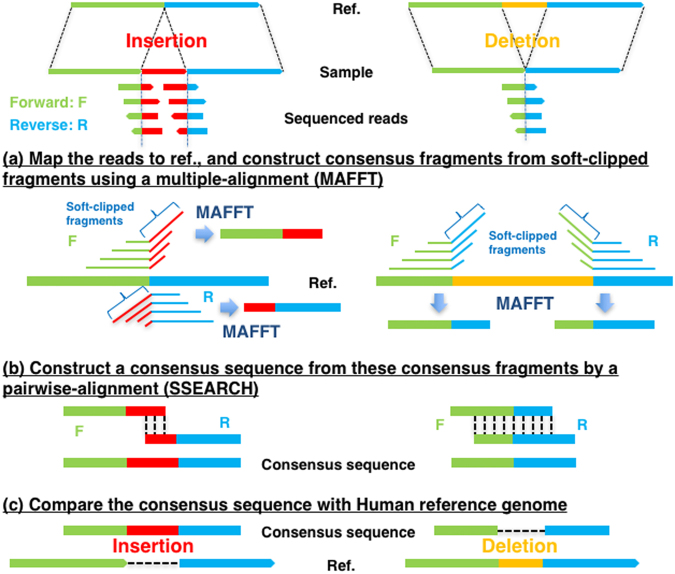
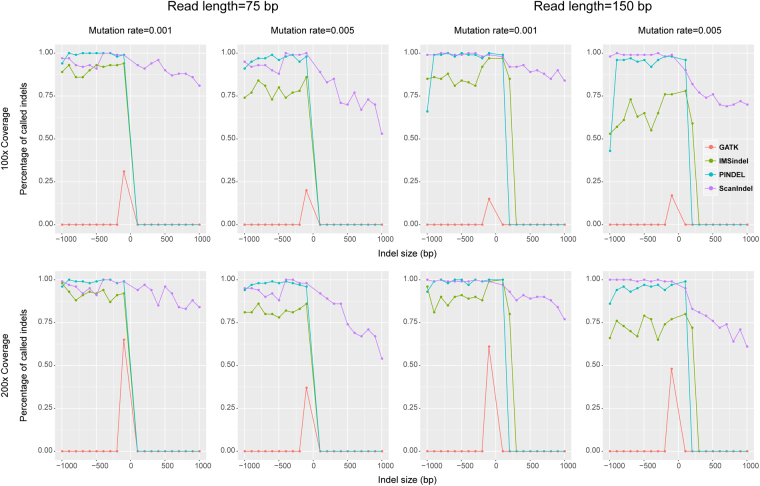
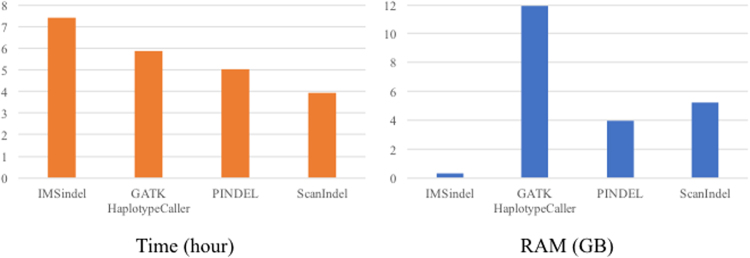
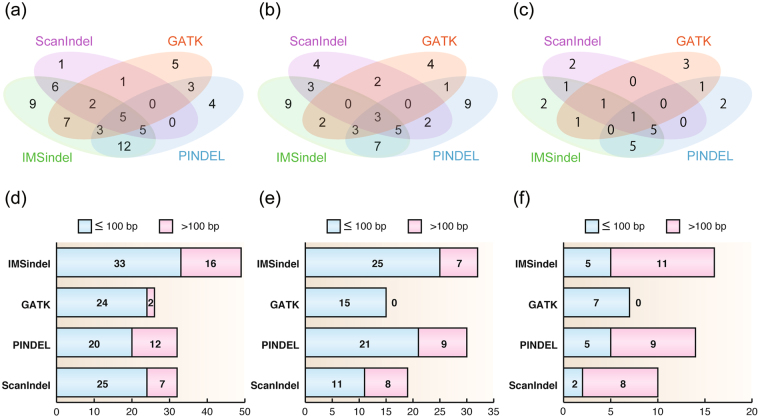
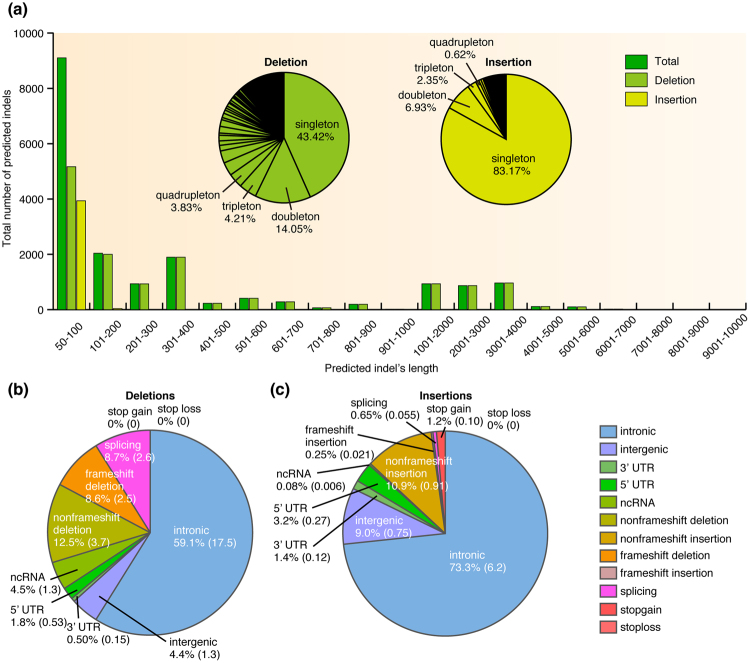
Insertions and deletions (indels) have been implicated in dozens of human diseases through the radical alteration of gene function by short frameshift indels as well as long indels. However, the accurate detection of these indels from next-generation sequencing data is still challenging. This is particularly true for intermediate-size indels (≥50 bp), due to the short DNA sequencing reads. Here, we developed a new method that predicts intermediate-size indels using BWA soft-clipped fragments (unmatched fragments in partially mapped reads) and unmapped reads. We report the performance comparison of our method, GATK, PINDEL and ScanIndel, using whole exome sequencing data from the same samples. False positive and false negative counts were determined through Sanger sequencing of all predicted indels across these four methods. The harmonic mean of the recall and precision, F-measure, was used to measure the performance of each method. Our method achieved the highest F-measure of 0.84 in one sample, compared to 0.56 for GATK, 0.52 for PINDEL and 0.46 for ScanIndel. Similar results were obtained in additional samples, demonstrating that our method was superior to the other methods for detecting intermediate-size indels. We believe that this methodology will contribute to the discovery of intermediate-size indels associated with human disease.
插入和缺失(indels)通过短的移码 indels 以及长的 indels 对基因功能产生剧烈改变,从而与数十种人类疾病相关。然而,从下一代测序数据中准确检测这些 indels 仍然具有挑战性。对于中等大小的 indels(≥50 bp),由于 DNA 测序读段较短,情况更是如此。在这里,我们开发了一种新的方法,该方法使用 BWA 软剪辑片段(部分映射读取中的未匹配片段)和未映射读取来预测中等大小的 indels。我们报告了使用来自相同样本的全外显子组测序数据,对我们的方法、GATK、PINDEL 和 ScanIndel 进行的性能比较。通过对这四种方法预测的所有 indels 进行 Sanger 测序,确定了假阳性和假阴性计数。我们使用每个方法的召回率和精度的调和平均值(F-measure)来衡量性能。在一个样本中,我们的方法的 F-measure 达到了最高的 0.84,而 GATK 为 0.56,PINDEL 为 0.52,ScanIndel 为 0.46。在其他样本中也得到了类似的结果,这表明我们的方法在检测中等大小的 indels 方面优于其他方法。我们相信,这种方法将有助于发现与人类疾病相关的中等大小的 indels。