Centre for Molecular Informatics, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK.
Division of Preclinical Innovation, National Center for Advancing Translational Sciences, National Institutes of Health, 9800 Medical Center Drive, Rockville, MD, 20852, USA.
Malar J. 2018 Apr 11;17(1):160. doi: 10.1186/s12936-018-2294-5.
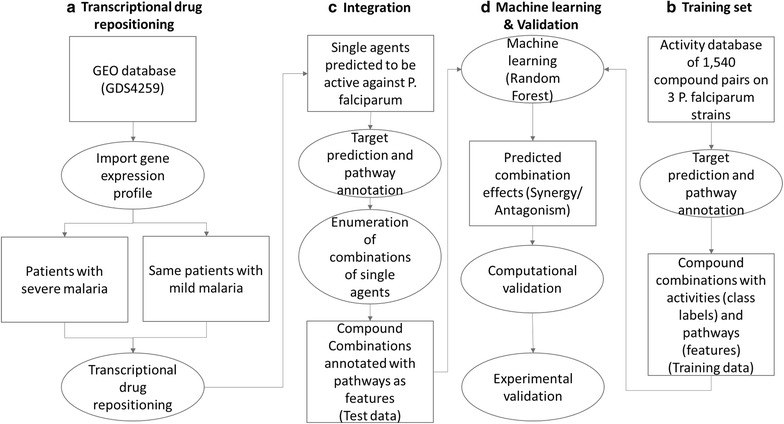
Nearly half of the world's population (3.2 billion people) were at risk of malaria in 2015, and resistance to current therapies is a major concern. While the standard of care includes drug combinations, there is a pressing need to identify new combinations that can bypass current resistance mechanisms. In the work presented here, a combined transcriptional drug repositioning/discovery and machine learning approach is proposed.
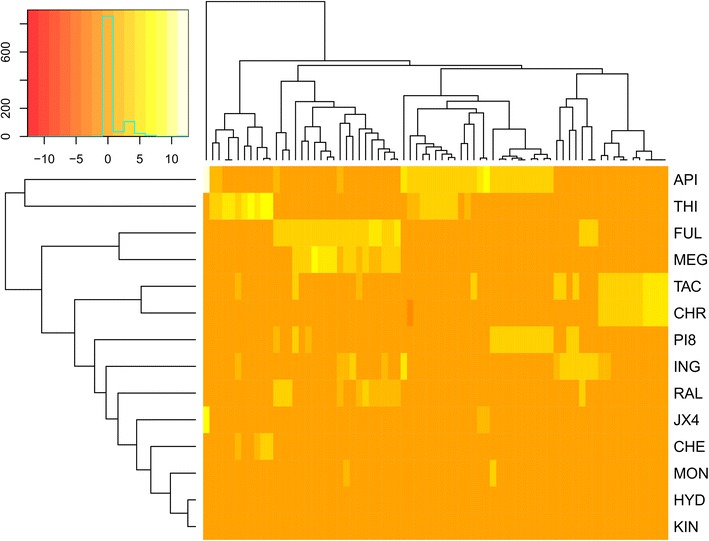
The integrated approach utilizes gene expression data from patient-derived samples, in combination with large-scale anti-malarial combination screening data, to predict synergistic compound combinations for three Plasmodium falciparum strains (3D7, DD2 and HB3). Both single compounds and combinations predicted to be active were prospectively tested in experiment.
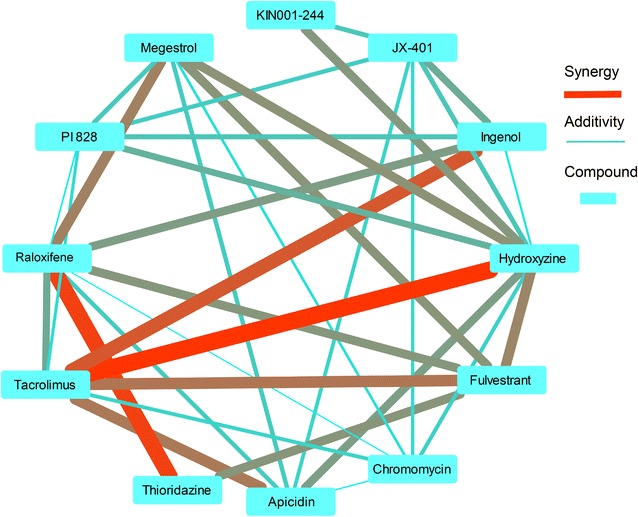
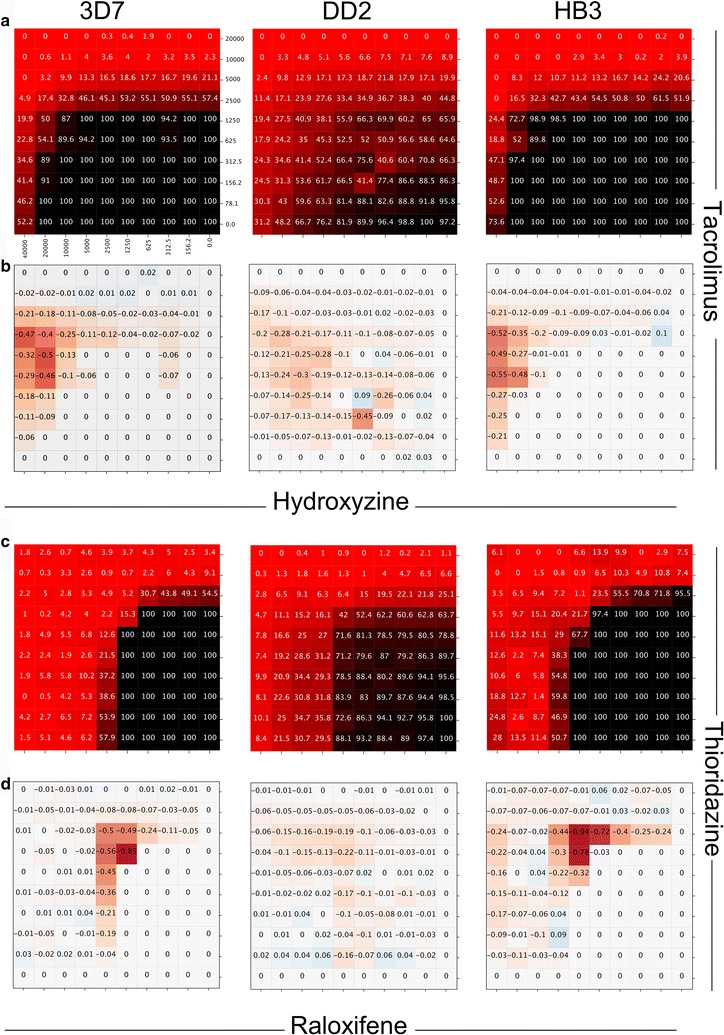
One of the predicted single agents, apicidin, was active with the AC50 values of 74.9, 84.1 and 74.9 nM in 3D7, DD2 and HB3 P. falciparum strains while its maximal safe plasma concentration in human is 547.6 ± 136.6 nM. Apicidin at the safe dose of 500 nM kills on average 97% of the parasite. The synergy prediction algorithm exhibited overall precision and recall of 83.5 and 65.1% for mild-to-strong, 48.8 and 75.5% for moderate-to-strong and 12.0 and 62.7% for strong synergies. Some of the prospectively predicted combinations, such as tacrolimus-hydroxyzine and raloxifene-thioridazine, exhibited significant synergy across the three P. falciparum strains included in the study.
Systematic approaches can play an important role in accelerating discovering novel combinational therapies for malaria as it enables selecting novel synergistic compound pairs in a more informed and cost-effective manner.
2015 年,全球近一半人口(32 亿人)面临疟疾风险,而对现有疗法的耐药性是一个主要关注点。虽然标准治疗包括药物联合治疗,但迫切需要确定可以绕过现有耐药机制的新联合治疗方案。在本文介绍的工作中,提出了一种结合转录药物重定位/发现和机器学习的方法。
该综合方法利用来自患者来源样本的基因表达数据,结合大规模抗疟联合筛选数据,预测三种恶性疟原虫(3D7、DD2 和 HB3)的协同化合物组合。预测具有活性的单一化合物和组合都在实验中进行了前瞻性测试。
预测的单一药物之一,apicidin,在 3D7、DD2 和 HB3 恶性疟原虫株中的 AC50 值分别为 74.9、84.1 和 74.9 nM,而在人体内的最大安全血浆浓度为 547.6±136.6 nM。apicidin 在安全剂量 500 nM 时平均杀死 97%的寄生虫。协同预测算法对轻度至重度协同作用的总体精度和召回率分别为 83.5%和 65.1%,对中度至重度协同作用的总体精度和召回率分别为 48.8%和 75.5%,对强协同作用的总体精度和召回率分别为 12.0%和 62.7%。一些前瞻性预测的组合,如他克莫司-羟嗪和雷洛昔芬-硫利达嗪,在研究中包括的三种恶性疟原虫株中表现出显著的协同作用。
系统性方法可以在加速发现疟疾新的联合治疗方案方面发挥重要作用,因为它可以以更明智和更具成本效益的方式选择新的协同化合物对。