Institute of Cancer and Genomic Sciences, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK.
Institute of Cancer and Genomic Sciences, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK; Sheffield Institute of Translational Neuroscience, University of Sheffield, Sheffield S10 2HQ, UK.
Exp Hematol. 2018 Jul;63:52-63.e5. doi: 10.1016/j.exphem.2018.04.002. Epub 2018 Apr 12.
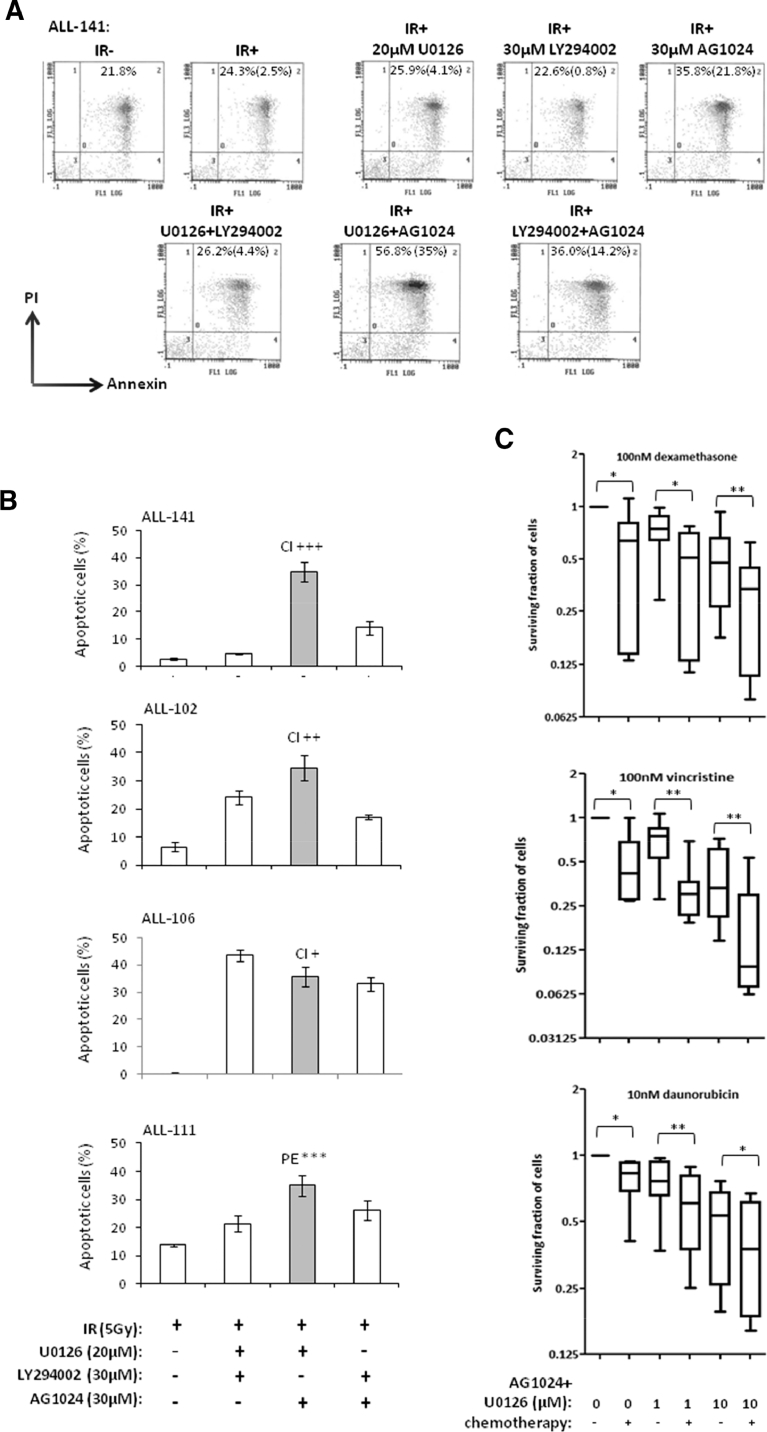
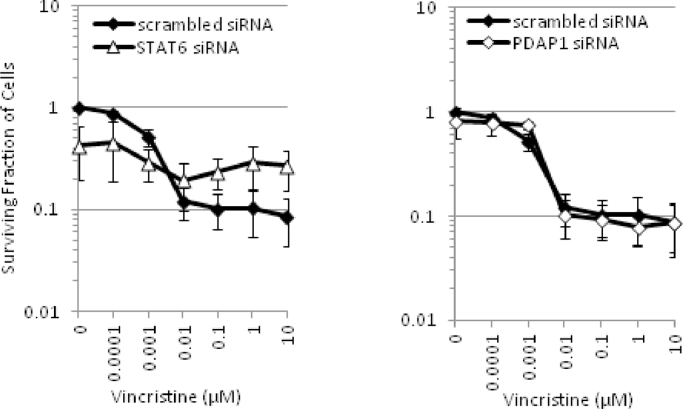
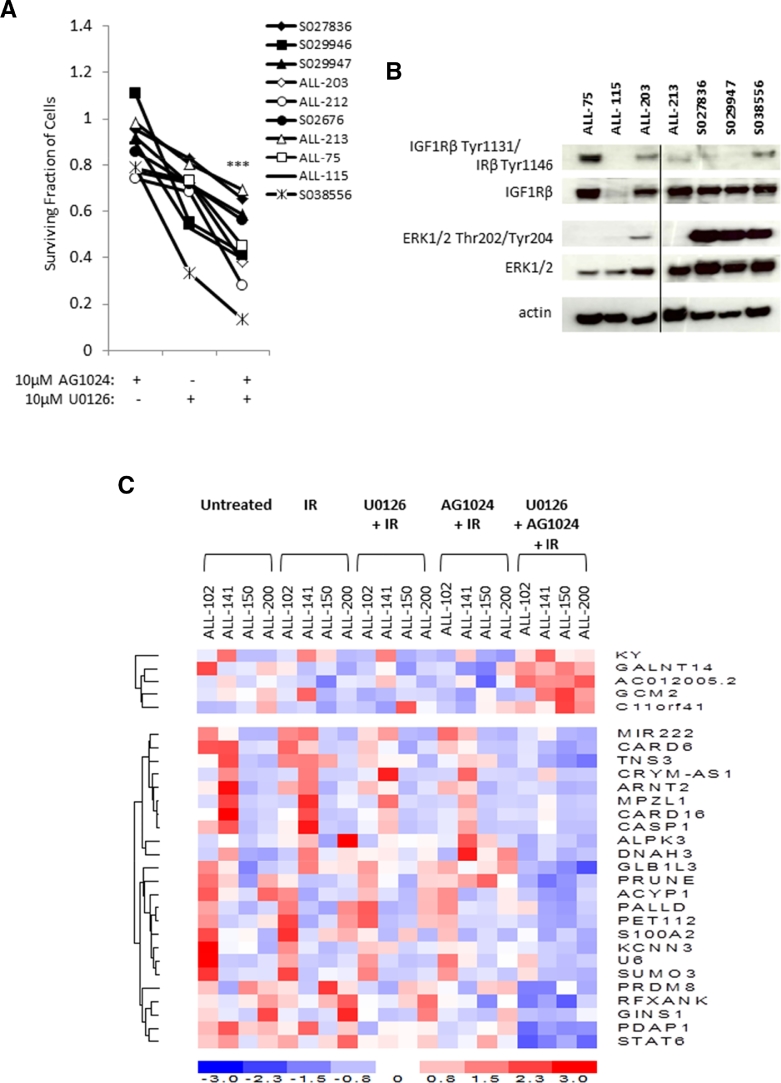
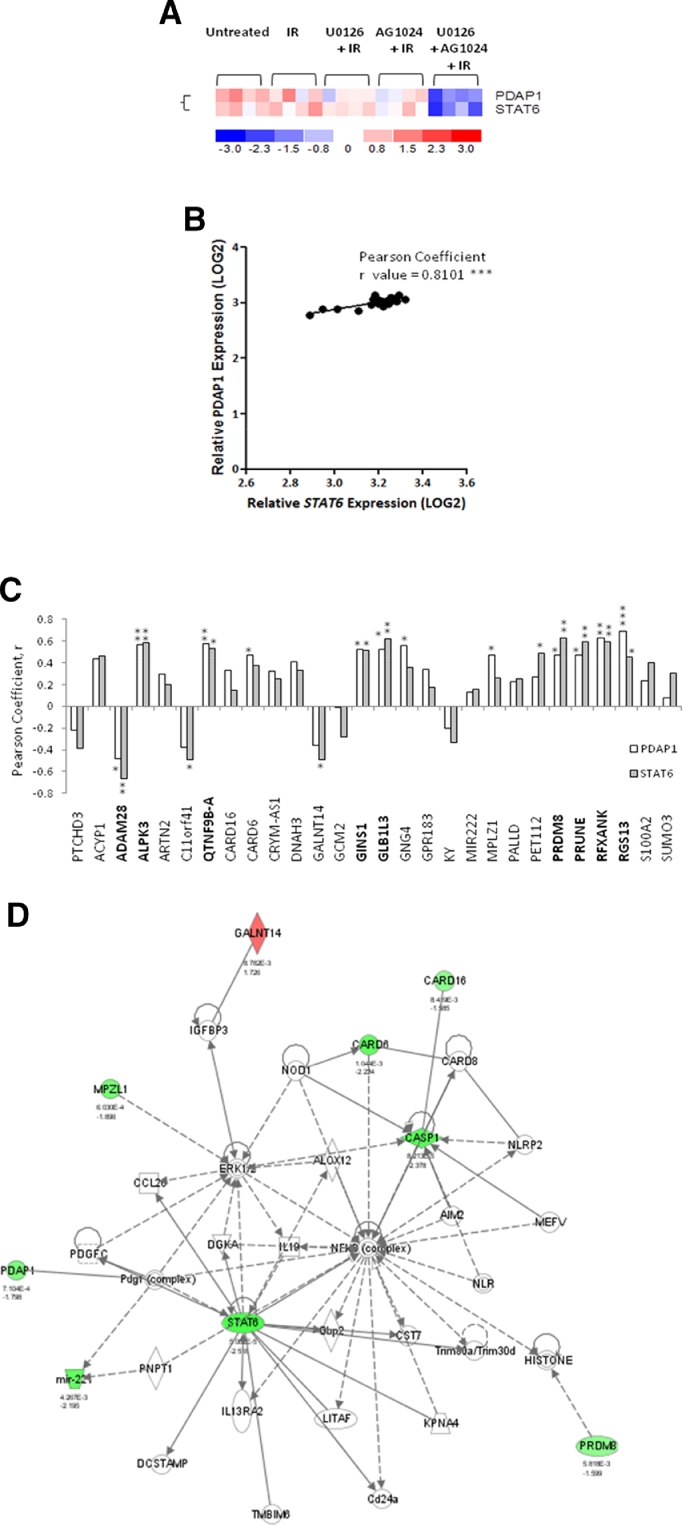
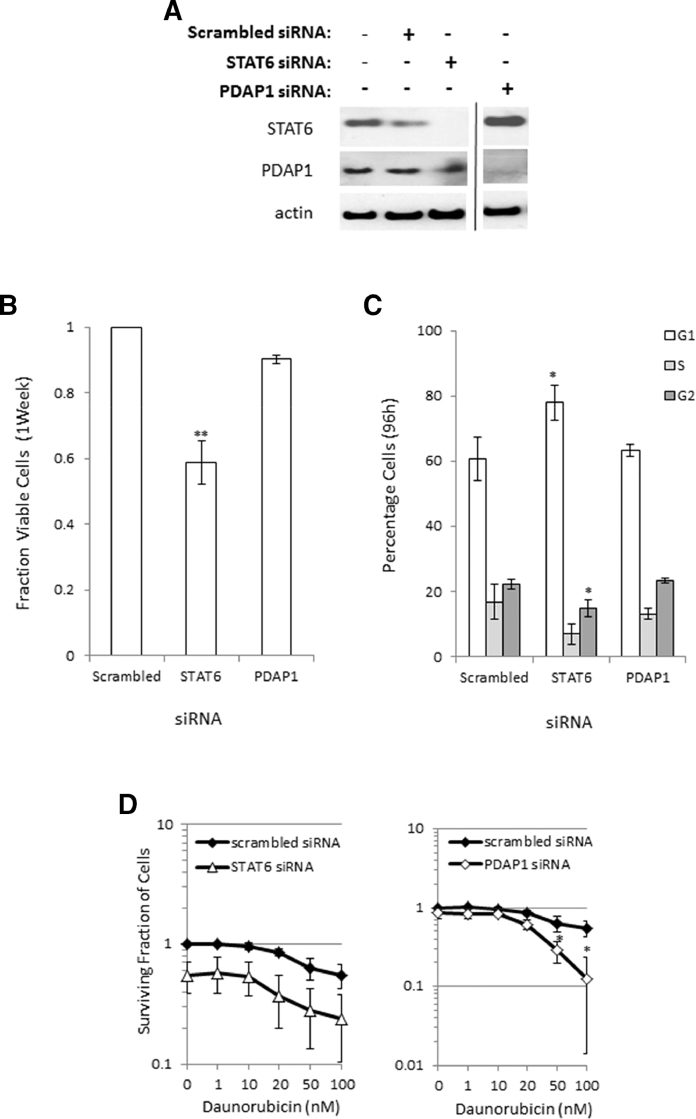
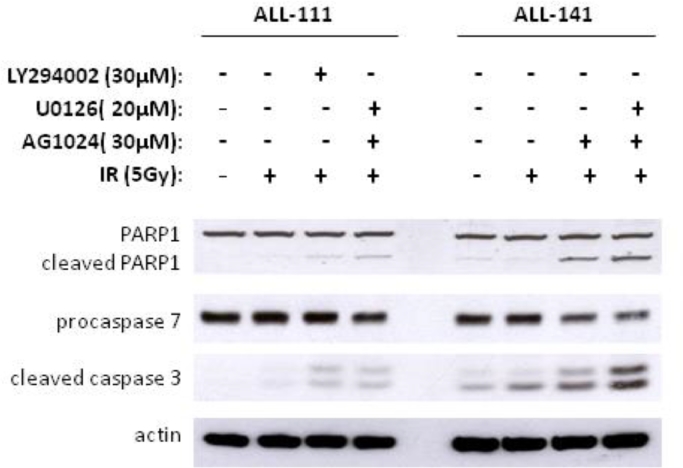
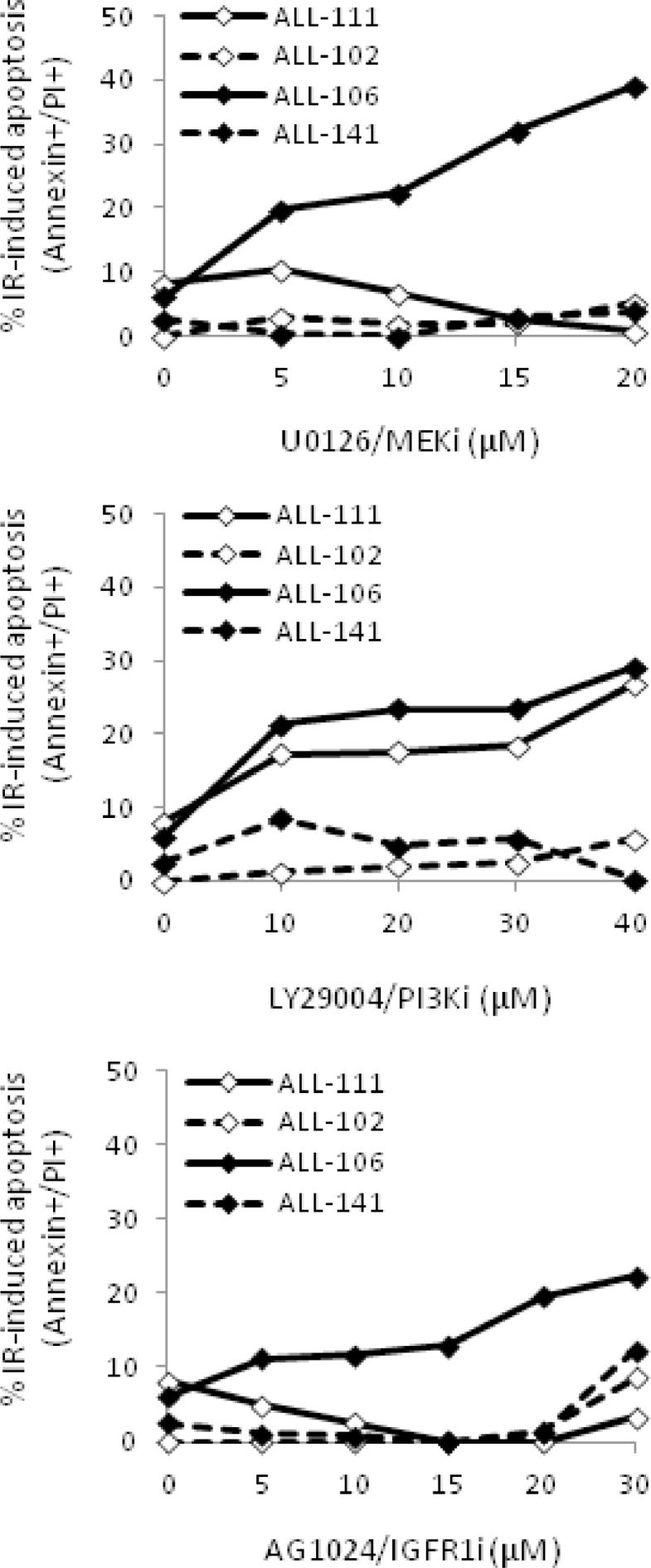
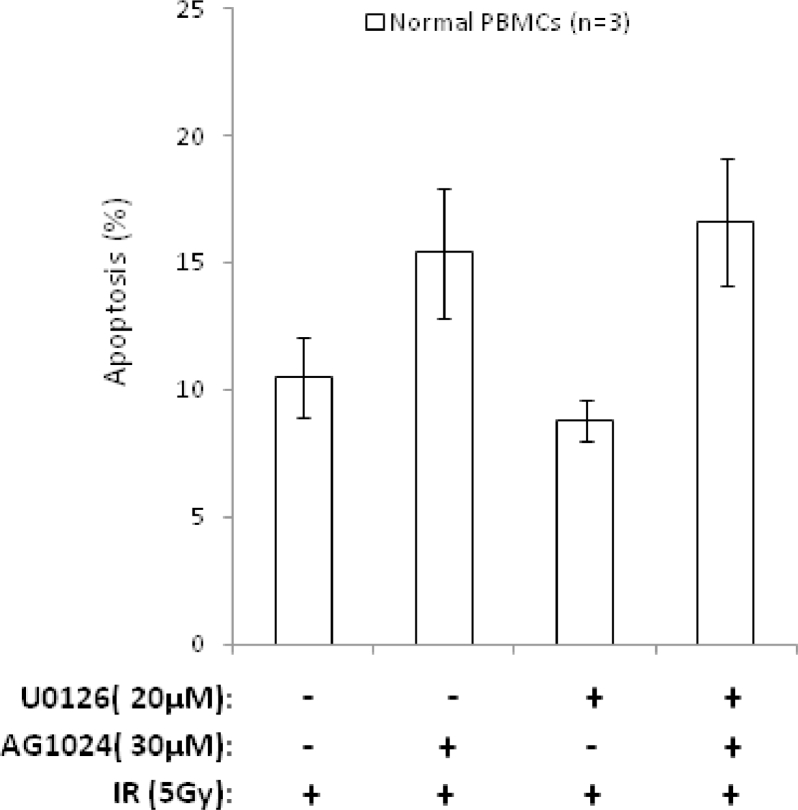
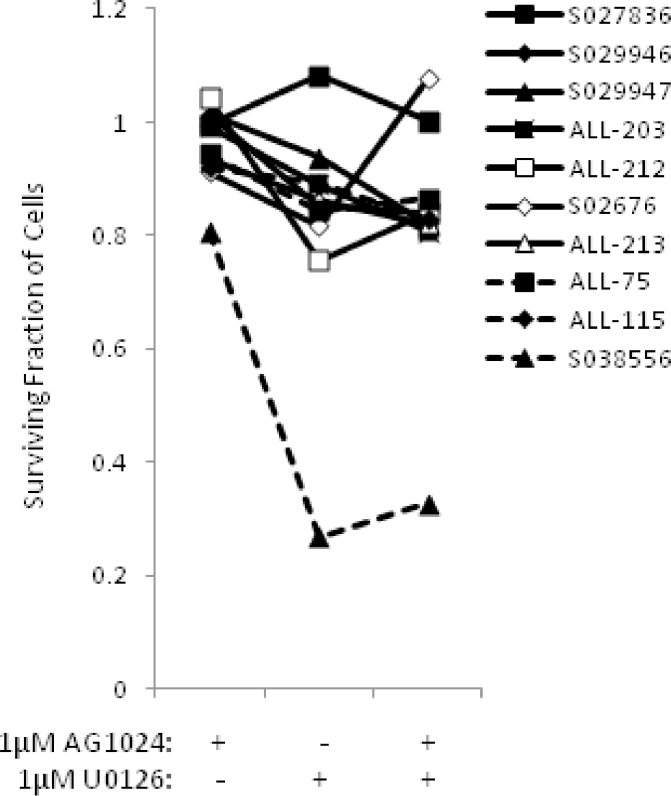
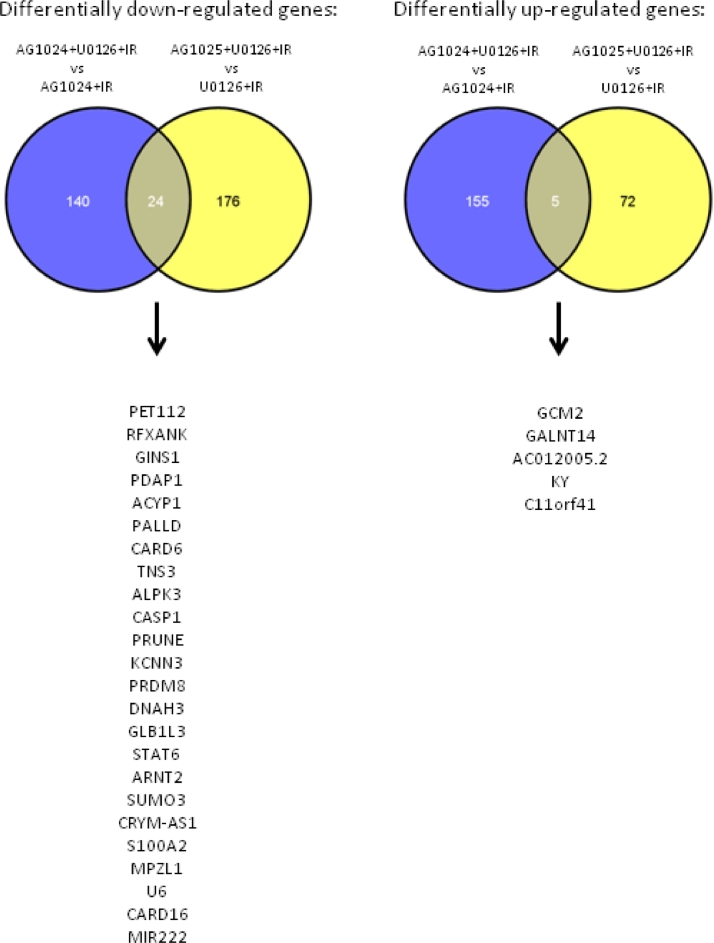
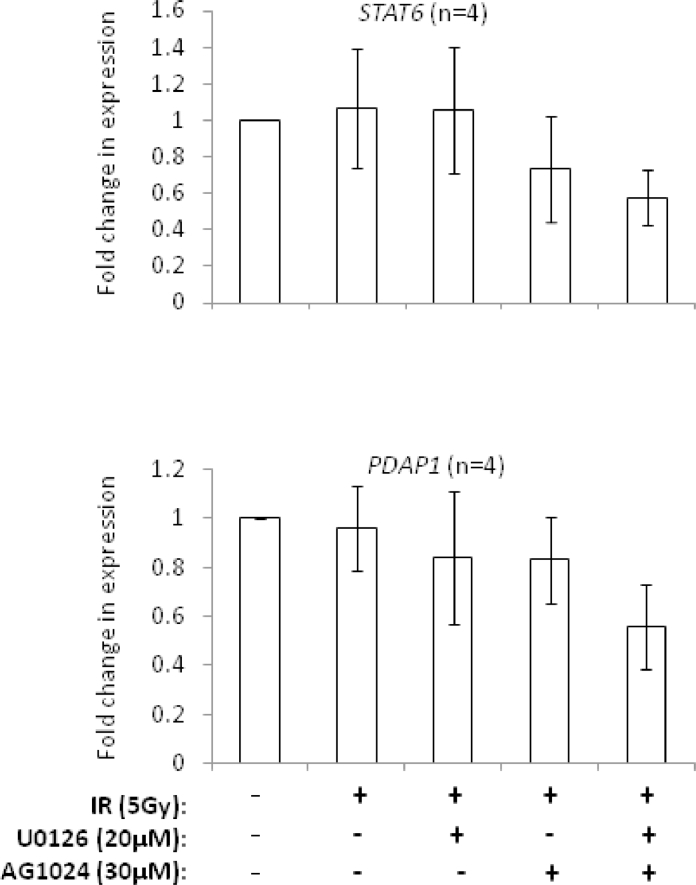
Heterogeneous upregulation of multiple prosurvival pathways underlies resistance to damage-induced apoptosis in acute lymphoblastic leukemia (ALL) cells despite normal p53 responses. Here, we show that the dual combination of insulin-like growth factor 1 (IGF1)/IGF1 receptor (IGF1/R) and mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (MEK) inhibition using AG1024 + U0126 can sensitize apoptosis-resistant ALL cells to ionizing radiation-induced DNA damage irrespective of effect of single pathway inhibition in vitro. This AG1024 + U0126 combination also significantly potentiates the ability of the core chemotherapy compounds vincristine, dexamethasone, and daunorubicin to kill ALL cells in vitro. Evidence of the synergistic action of AG1024 + U0126 in samples with variable basal levels of phosphorylated IGF1/Rβ and ERK1/2 suggested additional targets of this drug combination. Consistent with this, gene expression profiling identified 32 "synergy genes" differentially targeted by IGF1/R + MEK inhibition and, among these, Signal transducer and activator of transcription 6 (STAT6) and platelet-derived growth factor-associated protein 1 (PDAP1) were the most differentially downregulated cluster. Pearson correlation analysesrevealed that STAT6 and PDAP1 display significant expression codependency and a common expression pattern linked with other key "synergy" genes, supporting their predicted role in an STAT6-ERK-nuclear factor kappa beta (NF-κB) network. Knockdown studies revealed that loss of STAT6, but not PDAP1, impinges on the cell cycle, causing reduced numbers of viable cells. In combination with daunorubicin, STAT6 loss has an additive effect on cell killing, whereas PDAP1 loss is synergistic, indicating an important role of PDAP1 in the cellular response to this anthracycline. Inhibition of STAT6 or PDAP1 may therefore represent a potential novel therapeutic strategy for resistant ALL by enhancing sensitivity to chemotherapy.
尽管 p53 反应正常,但急性淋巴细胞白血病 (ALL) 细胞中多种抗凋亡途径的异质上调导致对损伤诱导的细胞凋亡产生抗性。在这里,我们表明,使用 AG1024+U0126 联合胰岛素样生长因子 1 (IGF1)/IGF1 受体 (IGF1/R) 和丝裂原活化蛋白激酶/细胞外信号调节激酶 (ERK) 激酶 (MEK) 抑制,可以使抗凋亡的 ALL 细胞对电离辐射诱导的 DNA 损伤敏感,而不管体外单一途径抑制的效果如何。这种 AG1024+U0126 联合用药还可以显著增强长春新碱、地塞米松和柔红霉素等核心化疗药物杀死 ALL 细胞的能力。在具有可变磷酸化 IGF1/Rβ 和 ERK1/2 基础水平的样本中,AG1024+U0126 联合用药具有协同作用的证据表明了这种药物联合的其他靶点。一致的是,基因表达谱分析确定了 32 个“协同基因”,这些基因被 IGF1/R+MEK 抑制靶向,其中信号转导和转录激活因子 6 (STAT6) 和血小板衍生生长因子相关蛋白 1 (PDAP1) 是下调最显著的簇。Pearson 相关分析显示,STAT6 和 PDAP1 显示出显著的表达相关性,具有与其他关键“协同”基因相关的共同表达模式,支持它们在 STAT6-ERK-核因子 kappa beta (NF-κB) 网络中的预测作用。敲低研究表明,STAT6 的缺失,但不是 PDAP1 的缺失,会影响细胞周期,导致活细胞数量减少。与柔红霉素联合使用时,STAT6 的缺失对细胞杀伤具有相加作用,而 PDAP1 的缺失则具有协同作用,表明 PDAP1 在细胞对这种蒽环类药物的反应中起着重要作用。因此,通过增强对化疗的敏感性,抑制 STAT6 或 PDAP1 可能代表一种抵抗 ALL 的潜在新治疗策略。