Bohmann Kristine, Gopalakrishnan Shyam, Nielsen Martin, Nielsen Luisa Dos Santos Bay, Jones Gareth, Streicker Daniel G, Gilbert M Thomas P
Section for Evolutionary Genomics, Natural History Museum of Denmark, University of Copenhagen, Copenhagen, Denmark.
School of Biological Sciences, University of East Anglia, Norwich, Norfolk, UK.
Mol Ecol Resour. 2018 Apr 19;18(5):1050-63. doi: 10.1111/1755-0998.12891.
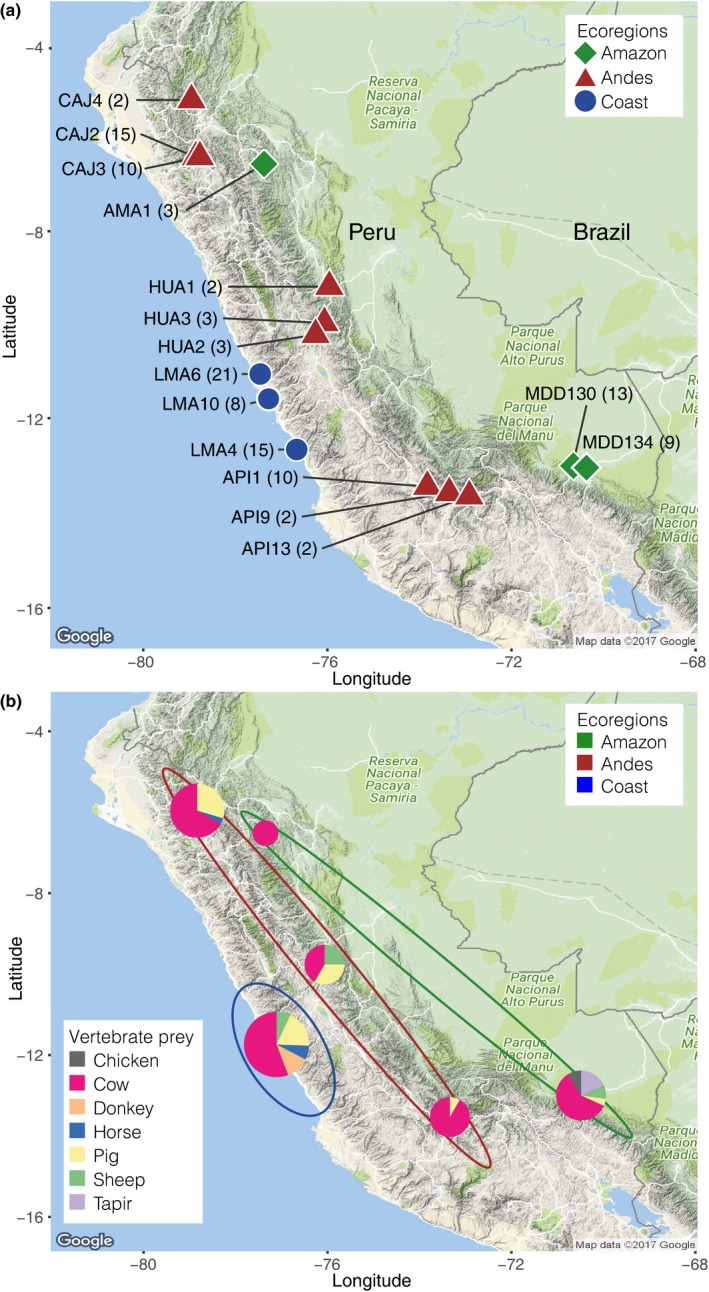
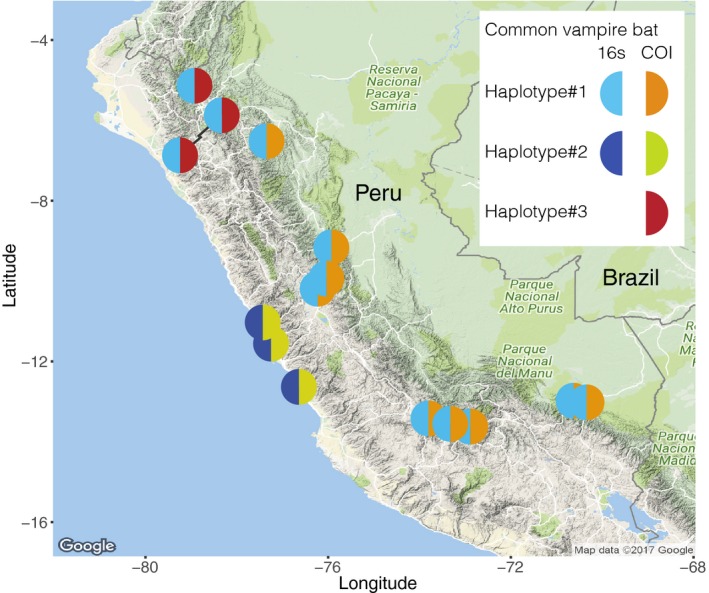
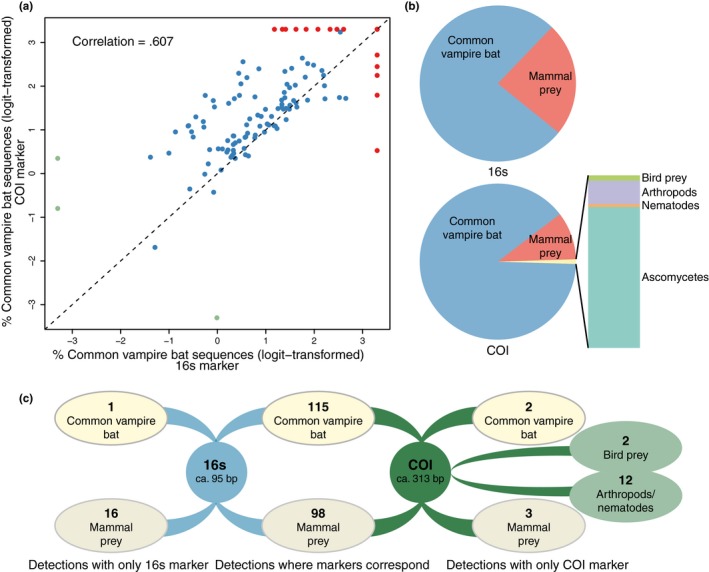
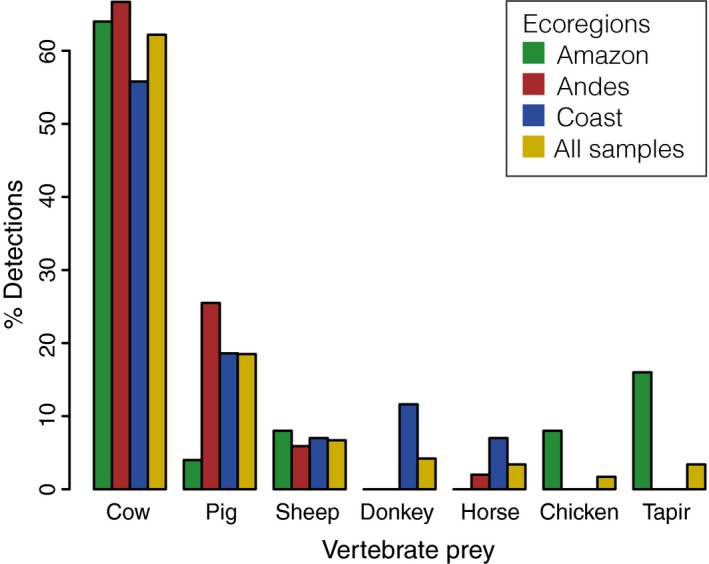
Metabarcoding diet analysis has become a valuable tool in animal ecology; however, co-amplified predator sequences are not generally used for anything other than to validate predator identity. Exemplified by the common vampire bat, we demonstrate the use of metabarcoding to infer predator population structure alongside diet assessments. Growing populations of common vampire bats impact human, livestock and wildlife health in Latin America through transmission of pathogens, such as lethal rabies viruses. Techniques to determine large-scale variation in vampire bat diet and bat population structure would empower locality- and species-specific projections of disease transmission risks. However, previously used methods are not cost-effective and efficient for large-scale applications. Using bloodmeal and faecal samples from common vampire bats from coastal, Andean and Amazonian regions of Peru, we showcase metabarcoding as a scalable tool to assess vampire bat population structure and feeding preferences. Dietary metabarcoding was highly effective, detecting vertebrate prey in 93.2% of the samples. Bats predominantly preyed on domestic animals, but fed on tapirs at one Amazonian site. In addition, we identified arthropods in 9.3% of samples, likely reflecting consumption of ectoparasites. Using the same data, we document mitochondrial geographic population structure in the common vampire bat in Peru. Such simultaneous inference of vampire bat diet and population structure can enable new insights into the interplay between vampire bat ecology and disease transmission risks. Importantly, the methodology can be incorporated into metabarcoding diet studies of other animals to couple information on diet and population structure.
代谢条形码饮食分析已成为动物生态学中的一种重要工具;然而,共同扩增出的捕食者序列通常除了用于验证捕食者身份外,并无其他用途。以普通吸血蝙蝠为例,我们展示了如何利用代谢条形码在进行饮食评估的同时推断捕食者的种群结构。普通吸血蝙蝠数量的不断增加,通过传播诸如致命狂犬病病毒等病原体,对拉丁美洲的人类、牲畜和野生动物健康造成影响。确定吸血蝙蝠饮食和蝙蝠种群结构大规模变化的技术,将有助于针对特定地点和物种预测疾病传播风险。然而,以前使用的方法对于大规模应用而言既不经济高效。我们利用来自秘鲁沿海、安第斯和亚马逊地区普通吸血蝙蝠的血餐和粪便样本,展示了代谢条形码作为一种可扩展工具,用于评估吸血蝙蝠的种群结构和摄食偏好。饮食代谢条形码分析非常有效,在93.2%的样本中检测到了脊椎动物猎物。蝙蝠主要捕食家畜,但在一个亚马逊地区的地点也捕食貘。此外,我们在9.3%的样本中鉴定出了节肢动物,这可能反映了对体外寄生虫的摄食。利用相同的数据,我们记录了秘鲁普通吸血蝙蝠的线粒体地理种群结构。这种对吸血蝙蝠饮食和种群结构的同时推断,能够为吸血蝙蝠生态学与疾病传播风险之间的相互作用带来新的见解。重要的是,该方法可纳入其他动物的代谢条形码饮食研究中,以结合饮食和种群结构方面的信息。