Dubowitz Neuromuscular Centre, UCL Great Ormond Street Institute of Child Health, London, United Kingdom.
Institute for Neuroscience and Muscle Research, Kid's Research Institute, Children's Hospital at Westmead, Sydney, New South Wales, Australia.
Ann Neurol. 2018 Jun;83(6):1105-1124. doi: 10.1002/ana.25241.
Comprehensive clinical characterization of congenital titinopathy to facilitate diagnosis and management of this important emerging disorder.
Using massively parallel sequencing we identified 30 patients from 27 families with 2 pathogenic nonsense, frameshift and/or splice site TTN mutations in trans. We then undertook a detailed analysis of the clinical, histopathological and imaging features of these patients.
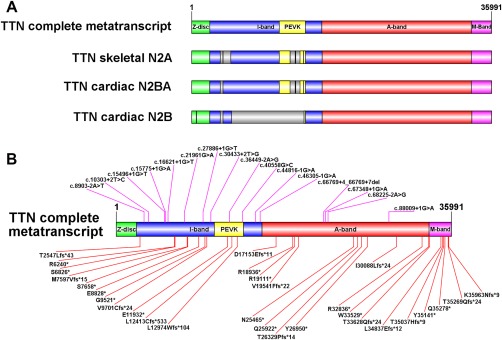
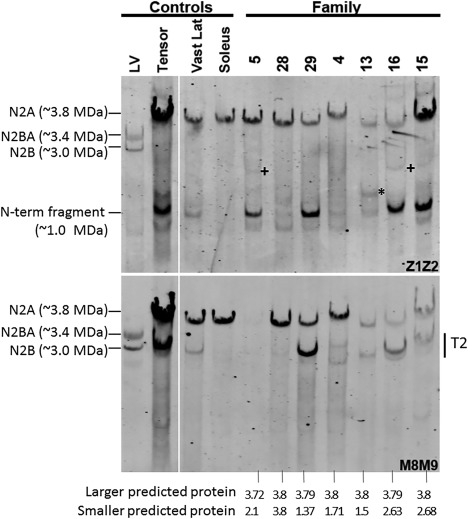
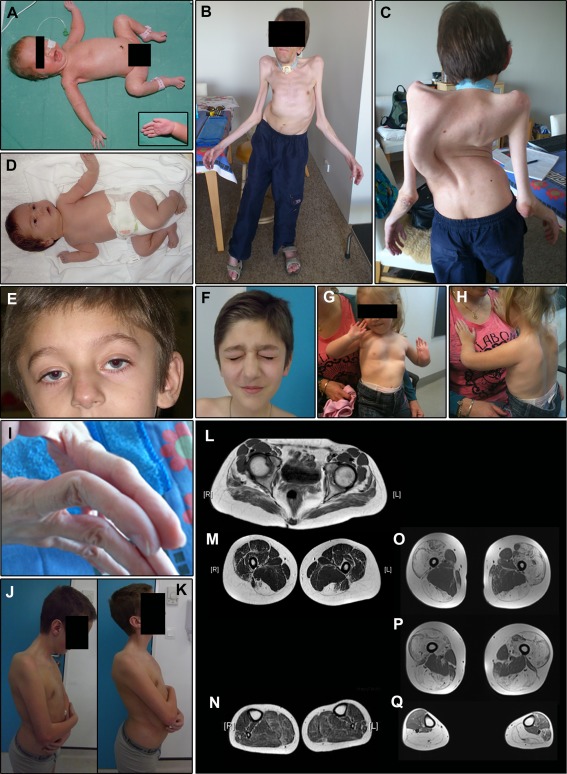
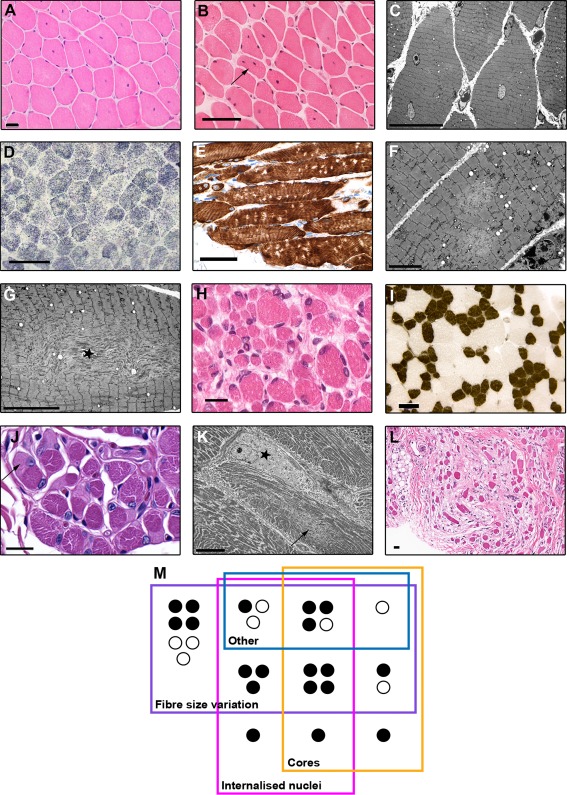
All patients had prenatal or early onset hypotonia and/or congenital contractures. None had ophthalmoplegia. Scoliosis and respiratory insufficiency typically developed early and progressed rapidly, whereas limb weakness was often slowly progressive, and usually did not prevent independent walking. Cardiac involvement was present in 46% of patients. Relatives of 2 patients had dilated cardiomyopathy. Creatine kinase levels were normal to moderately elevated. Increased fiber size variation, internalized nuclei and cores were common histopathological abnormalities. Cap-like regions, whorled or ring fibers, and mitochondrial accumulations were also observed. Muscle magnetic resonance imaging showed gluteal, hamstring and calf muscle involvement. Western blot analysis showed a near-normal sized titin protein in all samples. The presence of 2 mutations predicted to impact both N2BA and N2B cardiac isoforms appeared to be associated with greatest risk of cardiac involvement. One-third of patients had 1 mutation predicted to impact exons present in fetal skeletal muscle, but not included within the mature skeletal muscle isoform transcript. This strongly suggests developmental isoforms are involved in the pathogenesis of this congenital/early onset disorder.
This detailed clinical reference dataset will greatly facilitate diagnostic confirmation and management of patients, and has provided important insights into disease pathogenesis. Ann Neurol 2018;83:1105-1124.
全面临床描述先天性肌联蛋白病,以促进对此重要新兴疾病的诊断和管理。
我们使用大规模平行测序鉴定了 27 个家系的 30 名患者,这些家系的患者均携带 2 种致病性无义、移码和/或剪接位点 TTN 突变。然后,我们对这些患者的临床、组织病理学和影像学特征进行了详细分析。
所有患者均有产前或出生时即出现的肌无力和/或先天性挛缩。无眼外肌麻痹。脊柱侧凸和呼吸功能不全通常早期出现且快速进展,而肢体无力通常进展缓慢,通常不影响独立行走。46%的患者存在心脏受累。2 名患者的亲属患有扩张型心肌病。肌酸激酶水平正常或中度升高。纤维大小变异性增加、核内移和核内包涵体是常见的组织病理学异常。还观察到帽状区域、旋涡状或环状纤维以及线粒体堆积。肌肉磁共振成像显示臀肌、腘绳肌和小腿肌肉受累。Western blot 分析显示所有样本中的肌联蛋白蛋白大小接近正常。存在 2 种突变,预计同时影响 N2BA 和 N2B 型心脏同工型,似乎与心脏受累的最大风险相关。三分之一的患者有 1 种突变,预计影响胎儿骨骼肌中存在的外显子,但不包括成熟骨骼肌同工型转录本。这强烈表明发育同工型参与了这种先天性/早期发病疾病的发病机制。
此详细的临床参考数据集将极大地促进对患者的诊断确认和管理,并为疾病发病机制提供了重要的见解。