Anthropology and Human Genomics, Department of Biology II, Ludwig-Maximilians University, Grosshaderner Str. 2, 82152 Martinsried, Germany.
Gigascience. 2018 Jun 1;7(6). doi: 10.1093/gigascience/giy059.
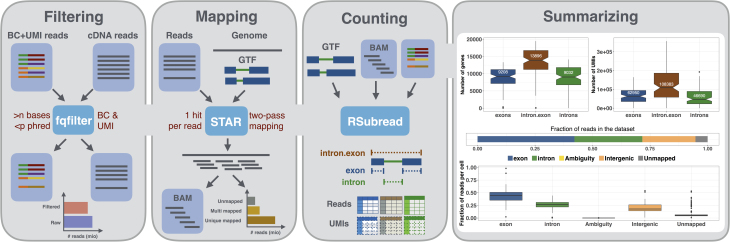
Single-cell RNA-sequencing (scRNA-seq) experiments typically analyze hundreds or thousands of cells after amplification of the cDNA. The high throughput is made possible by the early introduction of sample-specific bar codes (BCs), and the amplification bias is alleviated by unique molecular identifiers (UMIs). Thus, the ideal analysis pipeline for scRNA-seq data needs to efficiently tabulate reads according to both BC and UMI.
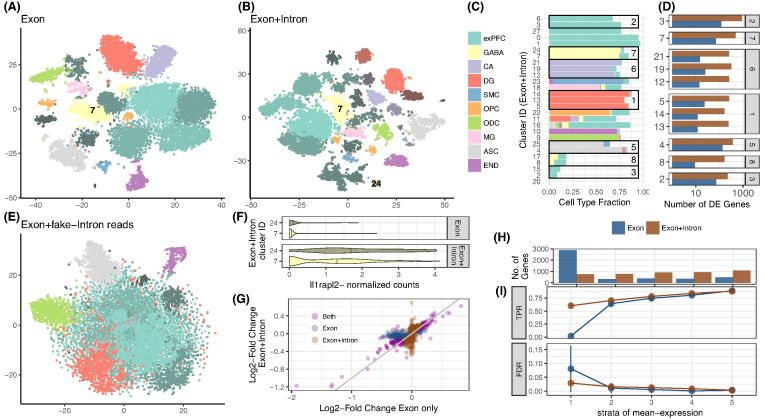
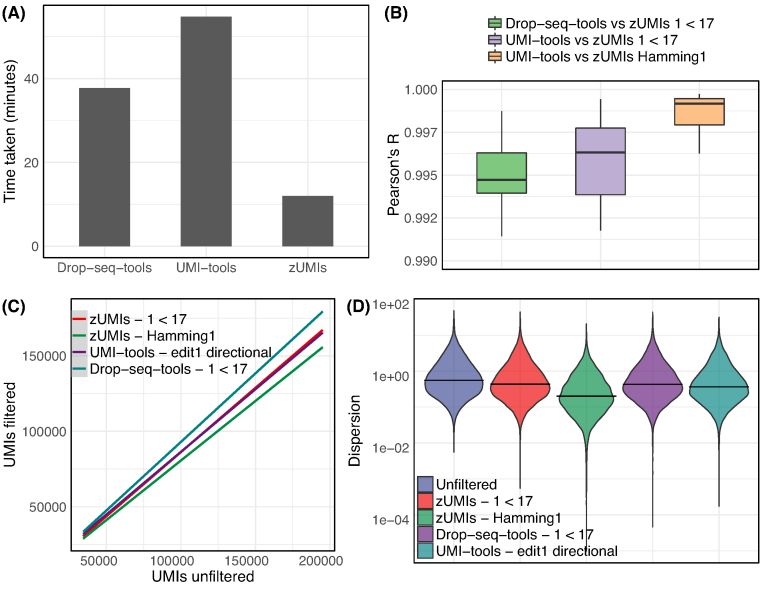
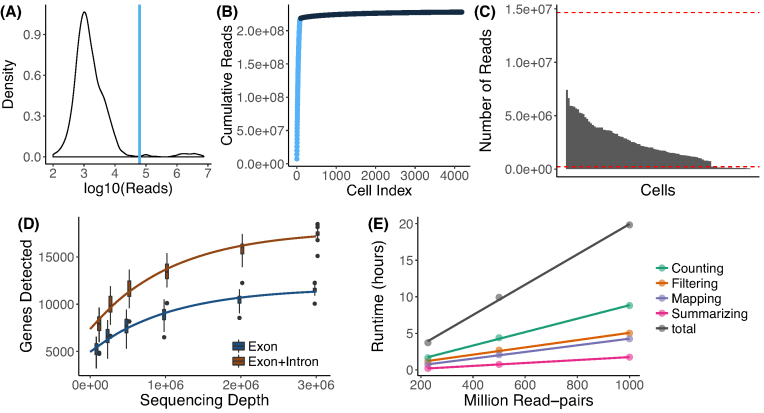
zUMIs is a pipeline that can handle both known and random BCs and also efficiently collapse UMIs, either just for exon mapping reads or for both exon and intron mapping reads. If BC annotation is missing, zUMIs can accurately detect intact cells from the distribution of sequencing reads. Another unique feature of zUMIs is the adaptive downsampling function that facilitates dealing with hugely varying library sizes but also allows the user to evaluate whether the library has been sequenced to saturation. To illustrate the utility of zUMIs, we analyzed a single-nucleus RNA-seq dataset and show that more than 35% of all reads map to introns. Also, we show that these intronic reads are informative about expression levels, significantly increasing the number of detected genes and improving the cluster resolution.
zUMIs flexibility makes if possible to accommodate data generated with any of the major scRNA-seq protocols that use BCs and UMIs and is the most feature-rich, fast, and user-friendly pipeline to process such scRNA-seq data.
单细胞 RNA 测序 (scRNA-seq) 实验通常在 cDNA 扩增后分析数百或数千个细胞。通过早期引入样本特异性条形码 (BC) 实现高通量,通过独特的分子标识符 (UMI) 缓解扩增偏差。因此,scRNA-seq 数据的理想分析流程需要根据 BC 和 UMI 有效地对读取进行制表。
zUMIs 是一个能够处理已知和随机 BC 的管道,并且还可以有效地合并 UMI,无论是用于外显子映射读取还是用于外显子和内含子映射读取。如果缺少 BC 注释,zUMIs 可以根据测序读取的分布准确检测完整的细胞。zUMIs 的另一个独特功能是自适应下采样功能,它便于处理差异极大的文库大小,同时允许用户评估文库是否已测序饱和。为了说明 zUMIs 的实用性,我们分析了一个单核 RNA-seq 数据集,结果表明超过 35%的所有读取都映射到内含子上。此外,我们还表明这些内含子读取可提供有关表达水平的信息,显著增加了检测到的基因数量,并提高了聚类分辨率。
zUMIs 的灵活性使其能够适应使用 BC 和 UMI 的任何主要 scRNA-seq 协议生成的数据,并且是处理此类 scRNA-seq 数据最具特色、最快、最用户友好的管道。