UCL Institute for Women's Health, Maternal and Fetal Medicine, Perinatal Brain Repair Group, London, WC1E 6HX, UK.
School of Pharmacy, University of Reading, Reading, RG6 6UA, UK.
J Physiol. 2018 Dec;596(23):6043-6062. doi: 10.1113/JP275649. Epub 2018 Jul 11.
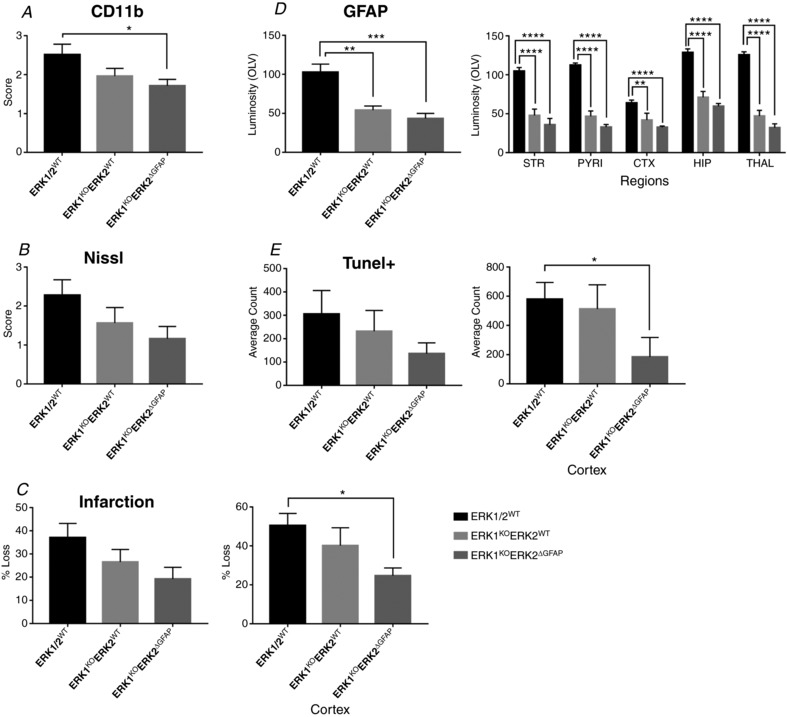
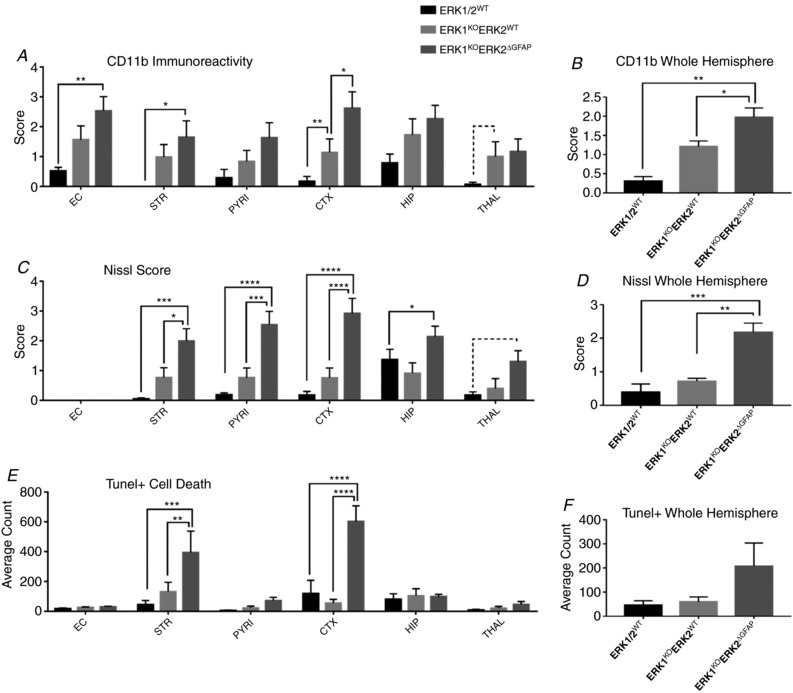
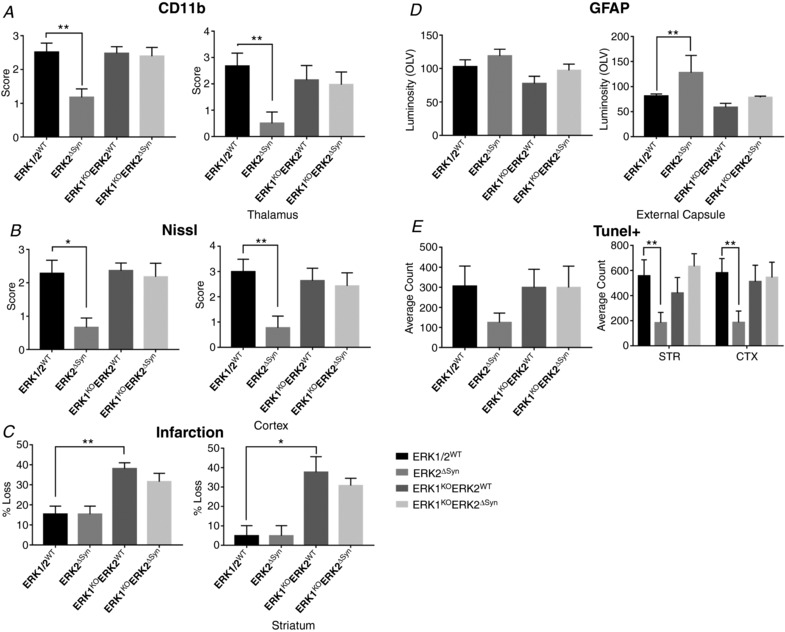
This study identifies phosphorylated extracellular signal-regulated kinase (ERK) to be immediately diminished followed by a rapid if transient increase for up to 4 h following hypoxic-ischaemic insult (HI) in the neonatal mouse. Phosphorylated ERK up-regulation was prevented with systemic injection of the mitogen-activated protein kinase kinase (MEK) inhibitor SL327. Treatment with SL327 both pre- and post-HI gave a strong reduction in the number of dying cells and microgliosis. By utilising transgenic mouse mutations, we observe that neuronal ERK2 significantly contributes to tissue damage, while ERK1 and astrocytic ERK2 are neuroprotective. Compared to global inactivation, selective cell-specific interference with ERK activity could result in stronger neuroprotection.
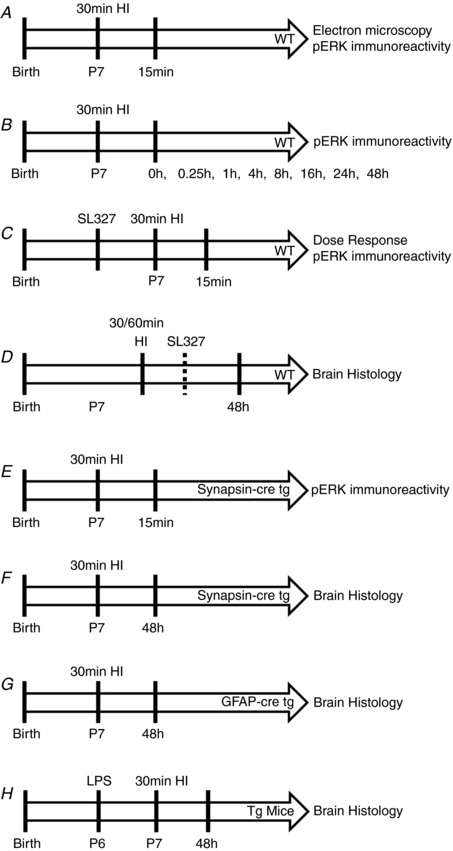
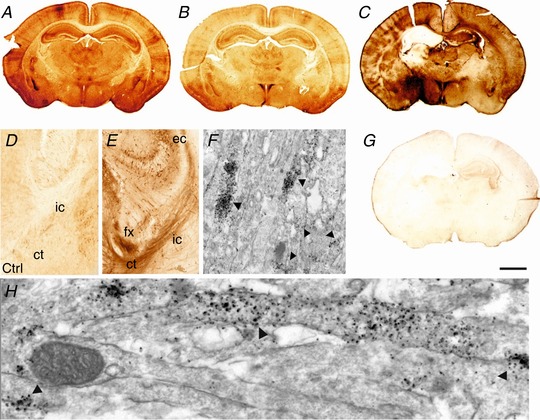
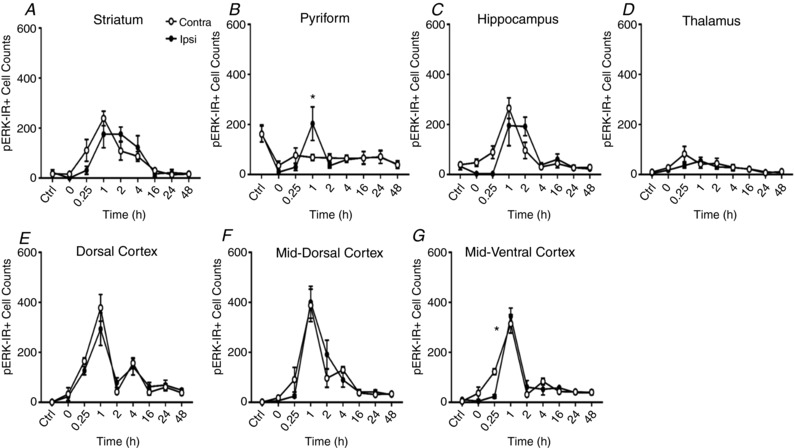
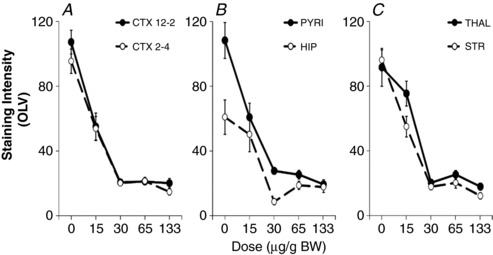
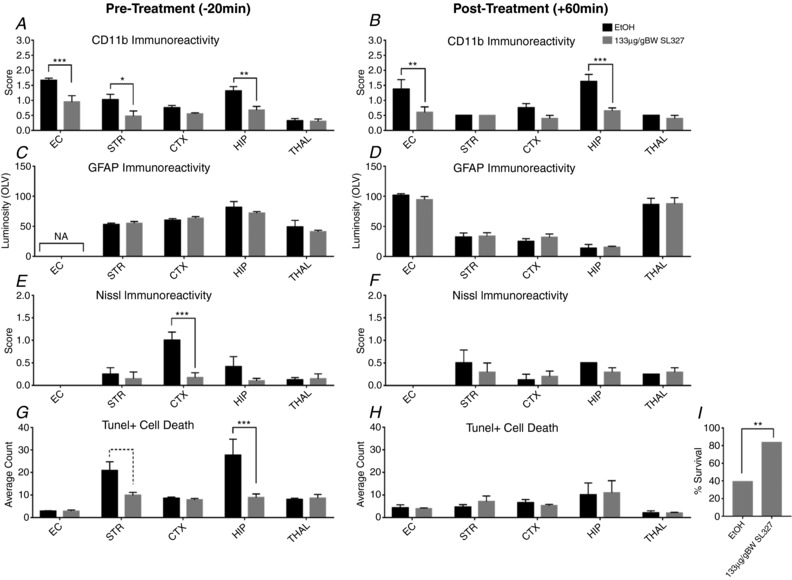
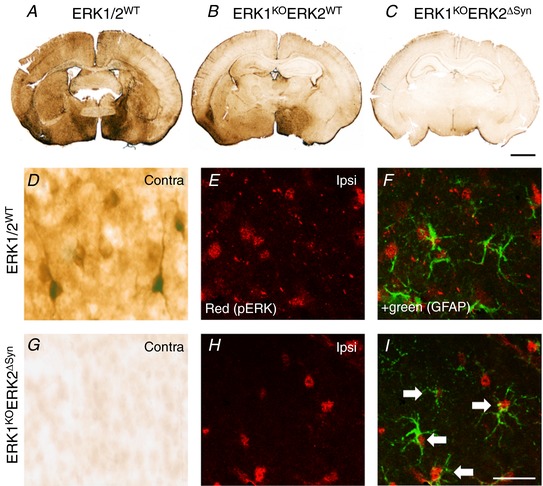
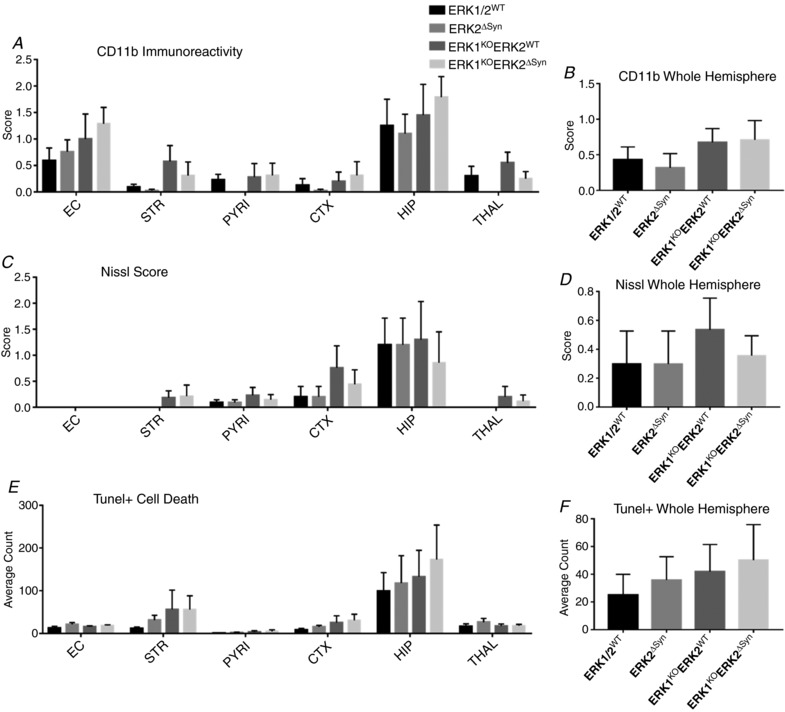
Hypoxia-ischaemia (HI) is a major cause of neonatal brain injury resulting in cerebral palsy, epilepsy, cognitive impairment and other neurological disabilities. The role of extracellular signal-regulated kinase (ERK) isoforms and their mitogen-activated protein kinase kinase (MEK)-dependent phosphorylation in HI has previously been explored but remains unresolved at cellular level. This is pertinent given the growing awareness of the role of non-neuronal cells in neuroprotection. Using a modified Rice-Vannucci model of HI in the neonatal mouse we observed time- and cell-dependent ERK phosphorylation (pERK), with strongly up-regulated pERK immunoreactivity first in periventricular white matter axons within 15-45 min of HI, followed by forebrain astrocytes and neurons (1-4 h post-HI), and return to baseline by 16 h. We explored the effects of pharmacological ERK blockade through the MEK inhibitor SL327 on neonatal HI-brain damage following HI alone (30 or 60 min) or lipopolysaccharide (LPS)-sensitised HI insult (30 min). Global inhibition of ERK phosphorylation with systemically applied SL327 abolished forebrain pERK immunoreactivity, and significantly reduced cell death and associated microglial activation at 48 h post-HI. We then explored the effects of cell-specific ERK2 deletion alone or in combination with global ERK1 knockout under the same conditions of HI insult. Neuronal ERK2 deletion strongly decreased infarct size, neuronal cell death and microglial activation in grey matter following both HI alone or LPS-sensitised HI. ERK1 deletion attenuated the protective effect of neuronal ERK2 deletion. Removal of astroglial ERK2 produced a reverse response, with a 3- to 4-fold increase in microglial activation and cell death. Our data suggest a cell-specific and time-dependent role of ERK in neonatal HI, with a predominant, neurotoxic effect of neuronal ERK2, which is counteracted by neuroprotection by ERK1 and astrocytic ERK2. Overall, global pharmacological inhibition of ERK phosphorylation is strongly neuroprotective.
本研究表明,在新生鼠缺氧缺血性损伤(HI)后,磷酸化细胞外信号调节激酶(ERK)立即减少,随后在 4 小时内迅速增加。通过全身注射丝裂原活化蛋白激酶激酶(MEK)抑制剂 SL327 可防止磷酸化 ERK 上调。HI 前和 HI 后给予 SL327 治疗均可显著减少死亡细胞和小胶质细胞的数量。通过利用转基因小鼠突变,我们观察到神经元 ERK2 显著导致组织损伤,而 ERK1 和星形胶质细胞 ERK2 具有神经保护作用。与全局失活相比,ERK 活性的选择性细胞特异性干扰可能会导致更强的神经保护作用。
缺氧缺血(HI)是导致脑瘫、癫痫、认知障碍和其他神经功能障碍的新生儿脑损伤的主要原因。ERK 同工型及其丝裂原活化蛋白激酶激酶(MEK)依赖性磷酸化在 HI 中的作用先前已被探索,但在细胞水平上仍未得到解决。鉴于非神经元细胞在神经保护中的作用日益受到重视,这一点尤为重要。本研究使用改良的 Rice-Vannucci 新生鼠 HI 模型,观察到 ERK 磷酸化(pERK)具有时间和细胞依赖性,HI 后 15-45 分钟内,脑室周围白质轴突中 pERK 免疫反应性强烈上调,随后是前脑星形胶质细胞和神经元(HI 后 1-4 小时),并在 16 小时内恢复基线。我们通过 MEK 抑制剂 SL327 研究了在单独 HI(30 或 60 分钟)或脂多糖(LPS)敏化 HI 损伤(30 分钟)后,ERK 药理学阻断对新生鼠 HI 脑损伤的影响。全身应用 SL327 抑制 ERK 磷酸化会消除前脑 pERK 免疫反应性,并在 HI 后 48 小时显著减少细胞死亡和相关小胶质细胞激活。然后,我们在相同的 HI 损伤条件下,单独或联合全局 ERK1 缺失,研究了细胞特异性 ERK2 缺失的影响。神经元 ERK2 缺失强烈减少了单独 HI 或 LPS 敏化 HI 后灰质中的梗死面积、神经元细胞死亡和小胶质细胞激活。ERK1 缺失减弱了神经元 ERK2 缺失的保护作用。去除星形胶质细胞 ERK2 导致小胶质细胞激活和细胞死亡增加 3-4 倍。我们的数据表明,ERK 在新生鼠 HI 中具有细胞特异性和时间依赖性作用,神经元 ERK2 具有主要的神经毒性作用,而 ERK1 和星形胶质细胞 ERK2 的神经保护作用可拮抗其作用。总体而言,ERK 磷酸化的全局药理学抑制具有强烈的神经保护作用。