Schlaffner Christoph N, Pirklbauer Georg J, Bender Andreas, Steen Judith A J, Choudhary Jyoti S
Department of Neurobiology, F. M. Kirby Neurobiology Center, Boston Children's Hospital, Harvard Medical School; Proteomic Mass Spectrometry, Wellcome Trust Sanger Institute, Wellcome Genome Campus; Centre for Molecular Informatics, Department of Chemistry, University of Cambridge;
Proteomic Mass Spectrometry, Wellcome Trust Sanger Institute, Wellcome Genome Campus.
J Vis Exp. 2018 May 22(135):57633. doi: 10.3791/57633.
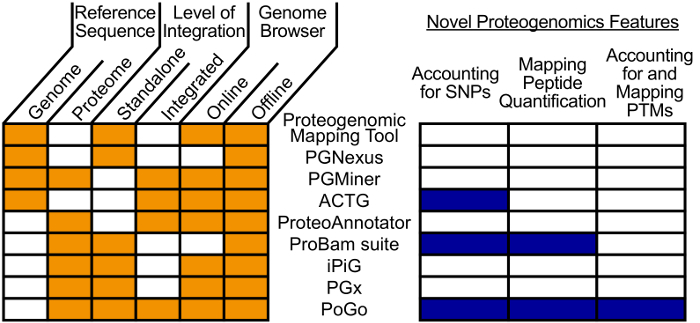
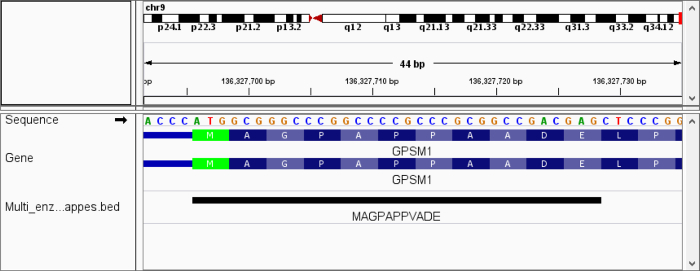
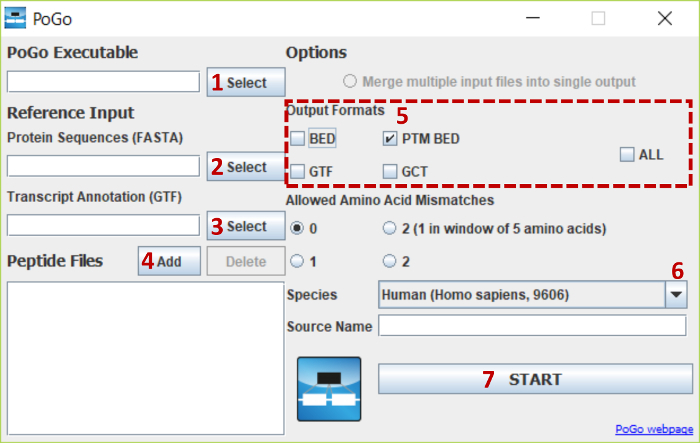
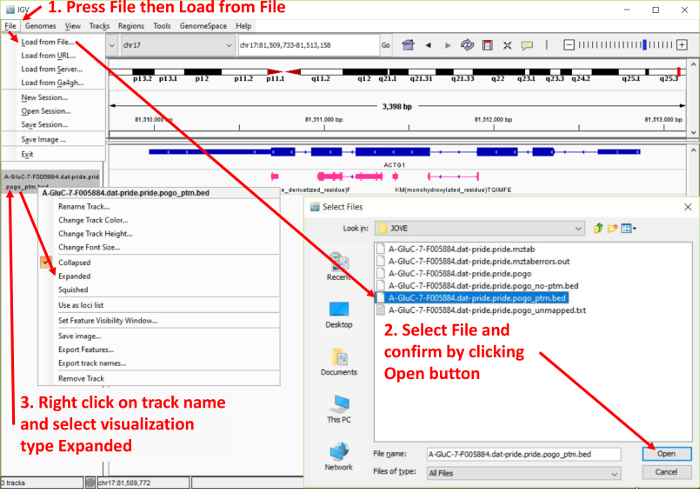
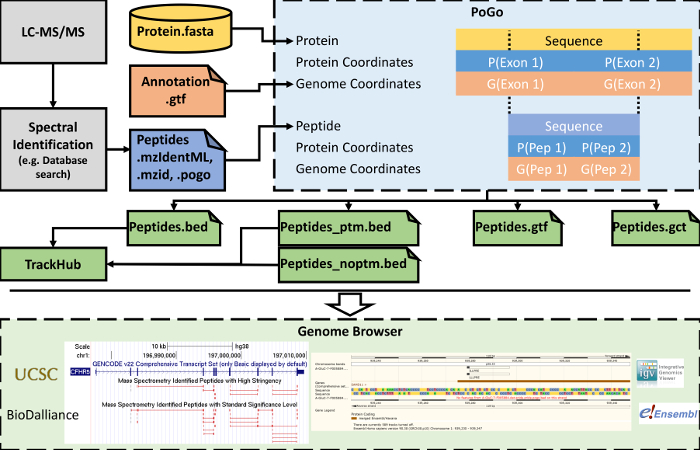
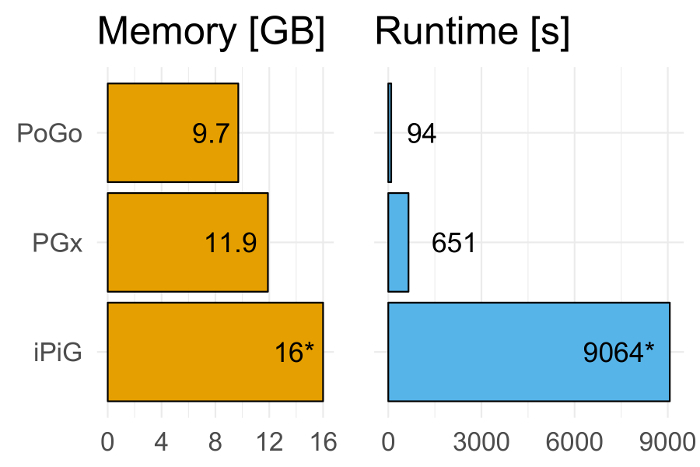
Cross-talk between genes, transcripts, and proteins is the key to cellular responses; hence, analysis of molecular levels as distinct entities is slowly being extended to integrative studies to enhance the understanding of molecular dynamics within cells. Current tools for the visualization and integration of proteomics with other omics datasets are inadequate for large-scale studies. Furthermore, they only capture basic sequence identify, discarding post-translational modifications and quantitation. To address these issues, we developed PoGo to map peptides with associated post-translational modifications and quantification to reference genome annotation. In addition, the tool was developed to enable the mapping of peptides identified from customized sequence databases incorporating single amino acid variants. While PoGo is a command line tool, the graphical interface PoGoGUI enables non-bioinformatics researchers to easily map peptides to 25 species supported by Ensembl genome annotation. The generated output borrows file formats from the genomics field and, therefore, visualization is supported in most genome browsers. For large-scale studies, PoGo is supported by TrackHubGenerator to create web-accessible repositories of data mapped to genomes that also enable an easy sharing of proteogenomics data. With little effort, this tool can map millions of peptides to reference genomes within only a few minutes, outperforming other available sequence-identity based tools. This protocol demonstrates the best approaches for proteogenomics mapping through PoGo with publicly available datasets of quantitative and phosphoproteomics, as well as large-scale studies.
基因、转录本和蛋白质之间的相互作用是细胞反应的关键;因此,将分子水平作为不同实体进行分析正逐渐扩展到整合研究,以加深对细胞内分子动力学的理解。目前用于蛋白质组学与其他组学数据集可视化和整合的工具不足以用于大规模研究。此外,它们仅捕获基本序列识别信息,而忽略了翻译后修饰和定量信息。为了解决这些问题,我们开发了PoGo,用于将具有相关翻译后修饰和定量信息的肽段映射到参考基因组注释上。此外,该工具还能够将从包含单氨基酸变体的定制序列数据库中鉴定出的肽段进行映射。虽然PoGo是一个命令行工具,但图形界面PoGoGUI使非生物信息学研究人员能够轻松地将肽段映射到Ensembl基因组注释支持的25种物种上。生成的输出采用了基因组学领域的文件格式,因此在大多数基因组浏览器中都支持可视化。对于大规模研究,TrackHubGenerator支持PoGo创建可通过网络访问的映射到基因组的数据存储库,这也便于蛋白质基因组学数据的轻松共享。只需付出很少的努力,该工具就能在几分钟内将数百万个肽段映射到参考基因组上,优于其他现有的基于序列同一性的工具。本方案展示了通过PoGo对公开可用的定量蛋白质组学和磷酸化蛋白质组学数据集以及大规模研究进行蛋白质基因组学映射的最佳方法。