Schiattarella Gabriele G, Boccella Nicola, Paolillo Roberta, Cattaneo Fabio, Trimarco Valentina, Franzone Anna, D'Apice Stefania, Giugliano Giuseppe, Rinaldi Laura, Borzacchiello Domenica, Gentile Alessandra, Lombardi Assunta, Feliciello Antonio, Esposito Giovanni, Perrino Cinzia
Department of Advanced Biomedical Sciences, University of Naples Federico II, Naples, Italy.
Department of Molecular Medicine and Medical Biotechnologies, University of Naples Federico II, Naples, Italy.
Front Physiol. 2018 May 28;9:558. doi: 10.3389/fphys.2018.00558. eCollection 2018.
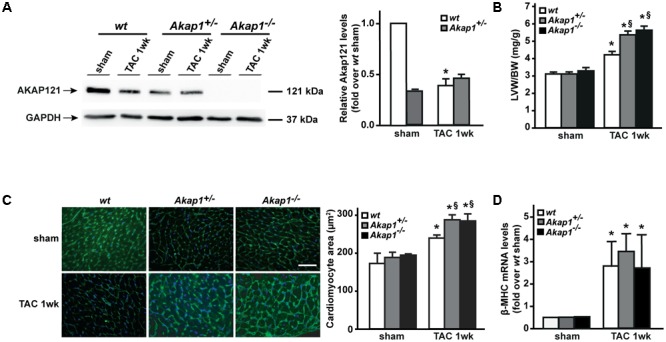
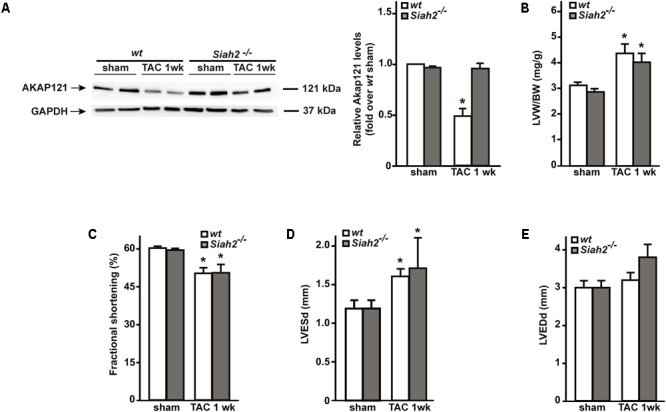
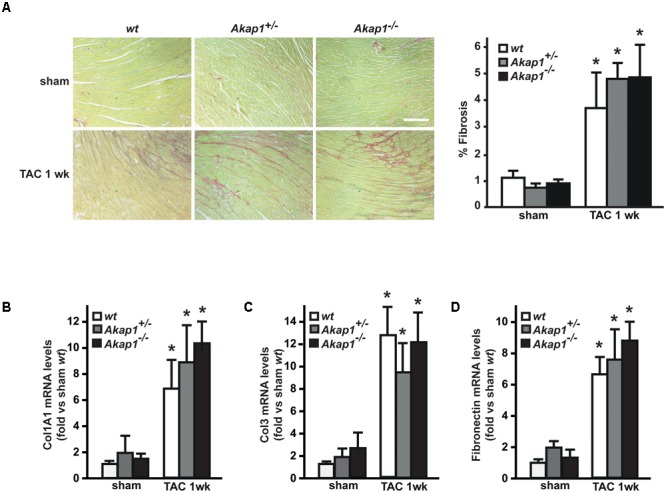
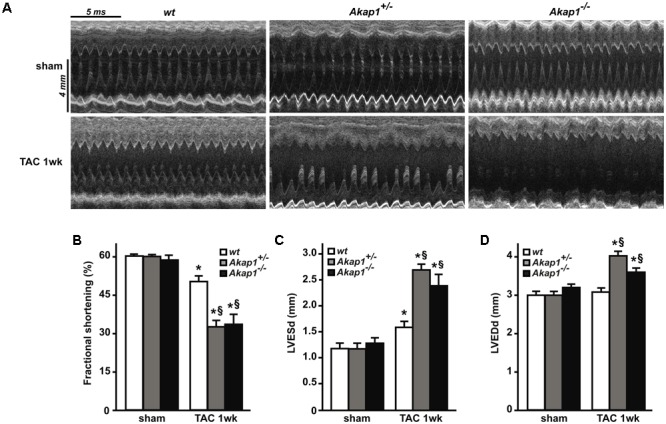
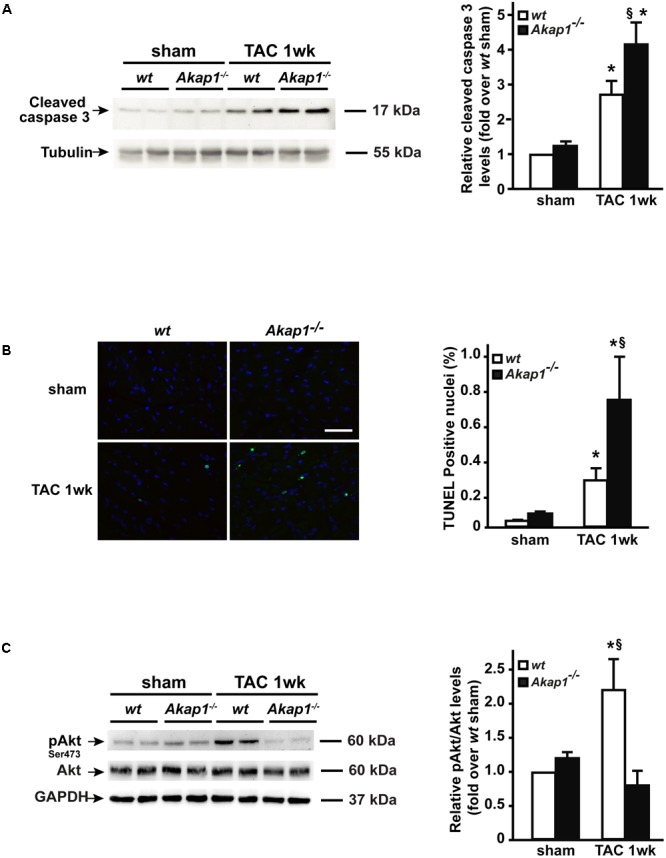
Left ventricular hypertrophy (LVH) is a major contributor to the development of heart failure (HF). Alterations in cyclic adenosine monophosphate (cAMP)-dependent signaling pathways participate in cardiomyocyte hypertrophy and mitochondrial dysfunction occurring in LVH and HF. cAMP signals are received and integrated by a family of cAMP-dependent protein kinase A (PKA) anchor proteins (AKAPs), tethering PKA to discrete cellular locations. AKAPs encoded by the gene (mitoAKAPs) promote PKA mitochondrial targeting, regulating mitochondrial structure and function, reactive oxygen species production, and cell survival. To determine the role of mitoAKAPs in LVH development, in the present investigation, mice with global genetic deletion of (), heterozygous (), and their wild-type () littermates underwent transverse aortic constriction (TAC) or SHAM procedure for 1 week. In mice, pressure overload induced the downregulation of AKAP121, the major cardiac mitoAKAP. Compared to mice did not display basal alterations in cardiac structure or function and cardiomyocyte size or fibrosis. However, loss of exacerbated LVH and cardiomyocyte hypertrophy induced by pressure overload and accelerated the progression toward HF in TAC mice, and these changes were not observed upon prevention of AKAP121 degradation in seven homolog 2 () knockout mice (). Loss of was also associated to a significant increase in cardiac apoptosis as well as lack of activation of Akt signaling after pressure overload. Taken together, these results demonstrate that genetic deletion of enhances LVH development and accelerates pressure overload-induced cardiac dysfunction, pointing at as a novel repressor of pathological LVH. These results confirm and extend the important role of mitoAKAPs in cardiac response to stress.
左心室肥厚(LVH)是心力衰竭(HF)发生发展的主要促成因素。环磷酸腺苷(cAMP)依赖性信号通路的改变参与了LVH和HF中发生的心肌细胞肥大和线粒体功能障碍。cAMP信号由cAMP依赖性蛋白激酶A(PKA)锚定蛋白(AKAPs)家族接收并整合,将PKA拴系到离散的细胞位置。由该基因编码的AKAPs(线粒体AKAPs)促进PKA向线粒体的靶向作用,调节线粒体结构和功能、活性氧生成以及细胞存活。为了确定线粒体AKAPs在LVH发生发展中的作用,在本研究中,对全局基因敲除()、杂合()的小鼠及其野生型()同窝小鼠进行了为期1周的横向主动脉缩窄(TAC)或假手术。在小鼠中,压力超负荷导致主要的心脏线粒体AKAP——AKAP121的下调。与小鼠相比,未表现出心脏结构或功能、心肌细胞大小或纤维化的基础改变。然而,基因缺失加剧了压力超负荷诱导的LVH和心肌细胞肥大,并加速了TAC小鼠向HF的进展,而在7种同源物2()基因敲除小鼠()中预防AKAP121降解后未观察到这些变化。基因缺失还与压力超负荷后心脏凋亡显著增加以及Akt信号未激活有关。综上所述,这些结果表明基因缺失增强了LVH的发展并加速了压力超负荷诱导的心脏功能障碍,表明是病理性LVH的一种新型抑制因子。这些结果证实并扩展了线粒体AKAPs在心脏应激反应中的重要作用。