John Walton Muscular Dystrophy Research Centre, MRC Centre for Neuromuscular Diseases, Institute of Genetic Medicine, Newcastle University, Newcastle, UK.
Leibniz-Institut für Analytische Wissenschaften-ISAS e.V, Dortmund, Germany.
Hum Mol Genet. 2018 Sep 15;27(18):3218-3232. doi: 10.1093/hmg/ddy225.
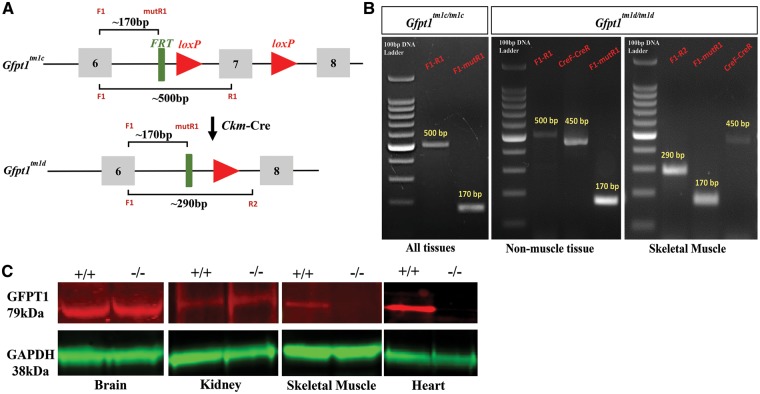
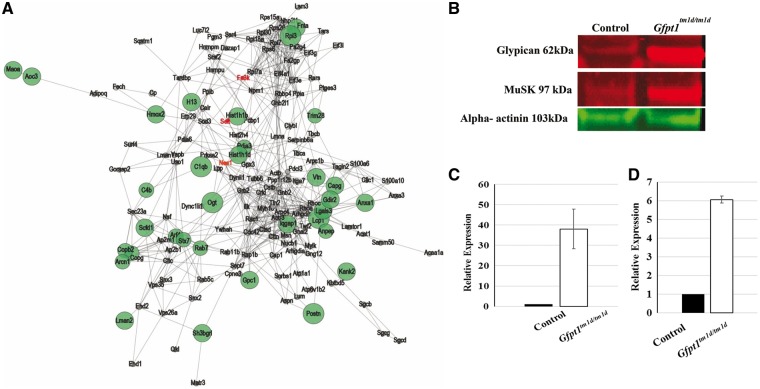
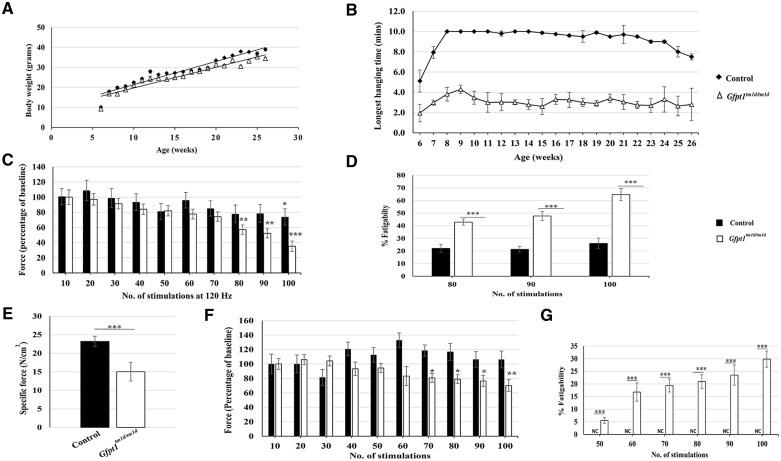
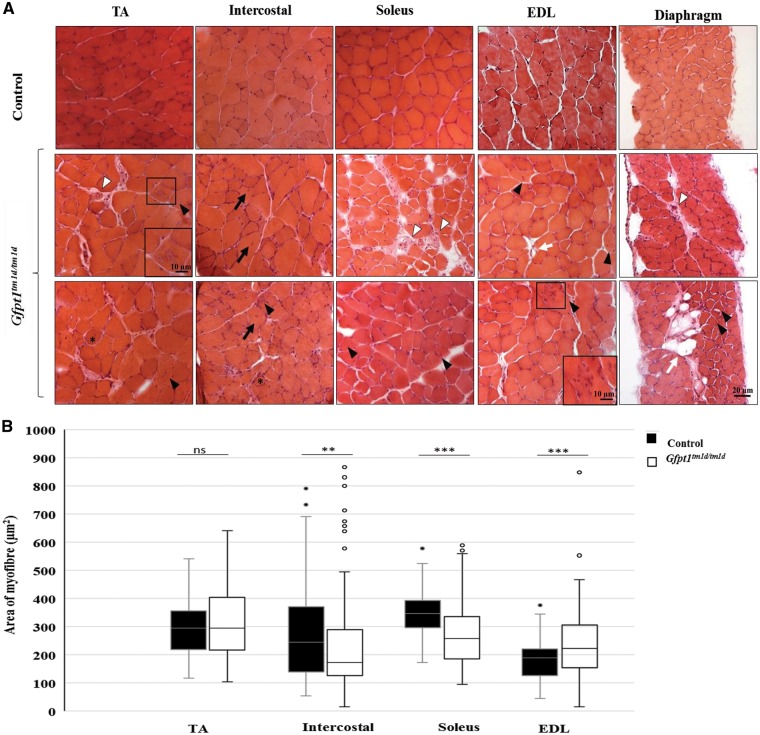
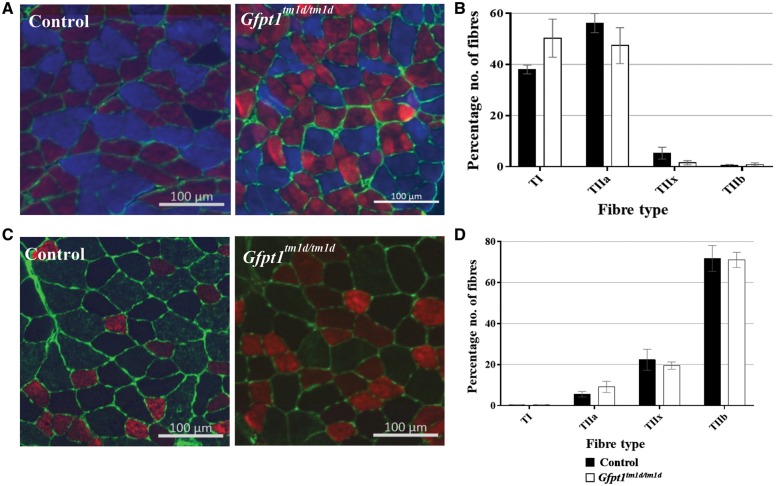
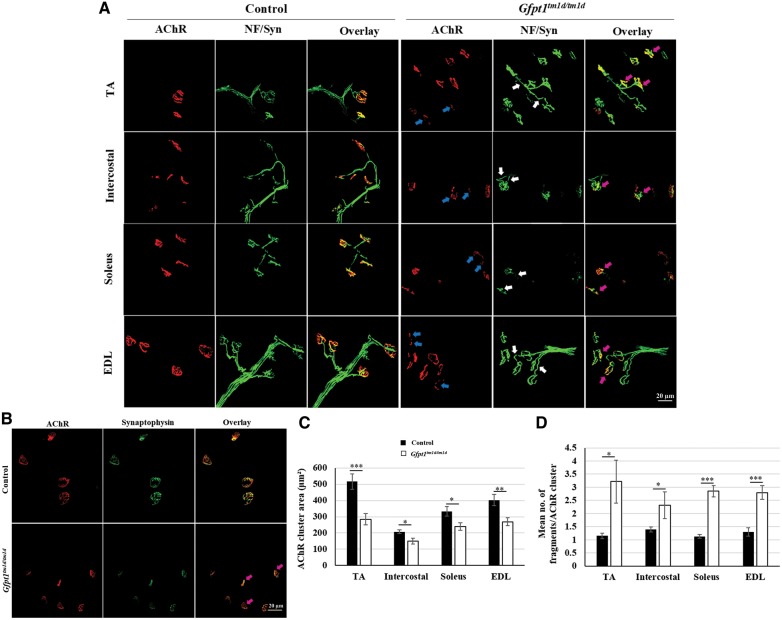
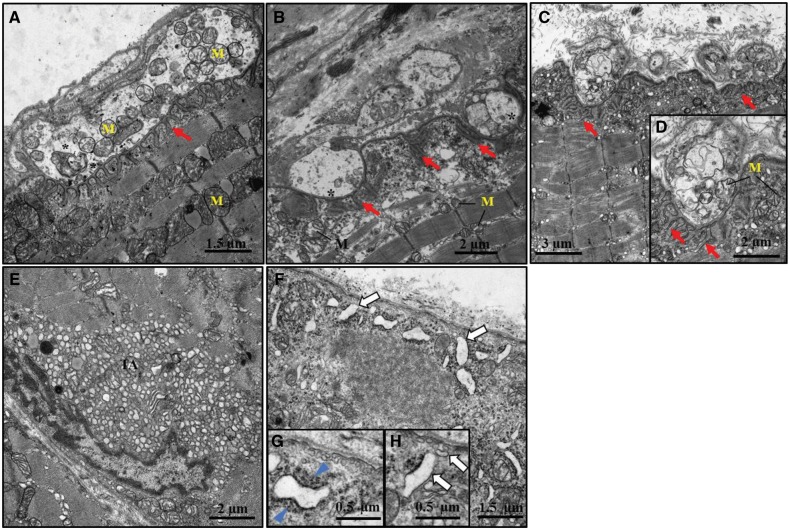
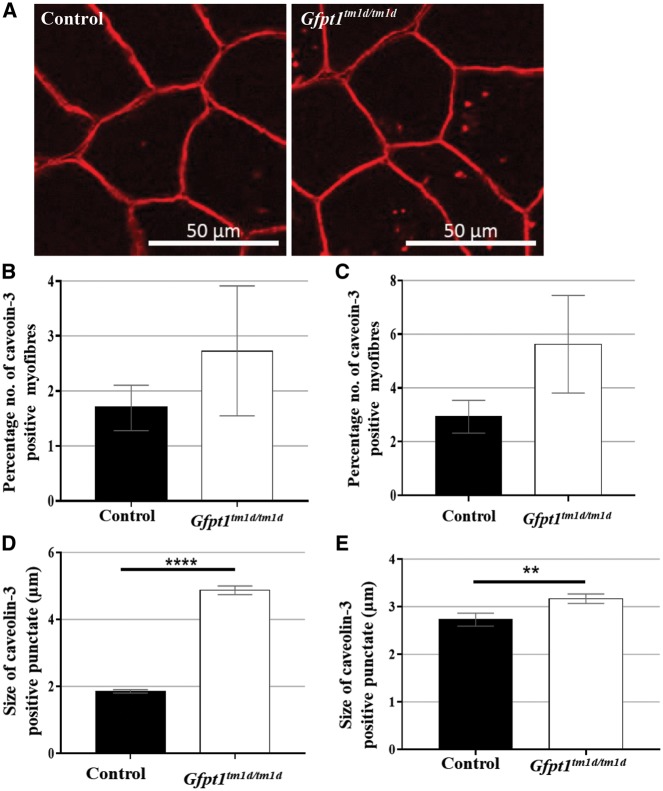
Glutamine-fructose-6-phosphate transaminase 1 (GFPT1) is the rate-limiting enzyme in the hexosamine biosynthetic pathway which yields precursors required for protein and lipid glycosylation. Mutations in GFPT1 and other genes downstream of this pathway cause congenital myasthenic syndrome (CMS) characterized by fatigable muscle weakness owing to impaired neurotransmission. The precise pathomechanisms at the neuromuscular junction (NMJ) owing to a deficiency in GFPT1 is yet to be discovered. One of the challenges we face is the viability of Gfpt1-/- knockout mice. In this study, we use Cre/LoxP technology to generate a muscle-specific GFPT1 knockout mouse model, Gfpt1tm1d/tm1d, characteristic of the human CMS phenotype. Our data suggest a critical role for muscle derived GFPT1 in the development of the NMJ, neurotransmission, skeletal muscle integrity and highlight that a deficiency in skeletal muscle alone is sufficient to cause morphological postsynaptic NMJ changes that are accompanied by presynaptic alterations despite the conservation of neuronal GFPT1 expression. In addition to the conventional morphological NMJ changes and fatigable muscle weakness, Gfpt1tm1d/tm1d mice display a progressive myopathic phenotype with the presence of tubular aggregates in muscle, characteristic of the GFPT1-CMS phenotype. We further identify an upregulation of skeletal muscle proteins glypican-1, farnesyltransferase/geranylgeranyltransferase type-1 subunit α and muscle-specific kinase, which are known to be involved in the differentiation and maintenance of the NMJ. The Gfpt1tm1d/tm1d model allows for further investigation of pathophysiological consequences on genes and pathways downstream of GFPT1 likely to involve misglycosylation or hypoglycosylation of NMJs and muscle targets.
谷氨酰胺果糖-6-磷酸转氨酶 1(GFPT1)是己糖胺生物合成途径中的限速酶,该途径产生蛋白质和脂质糖基化所需的前体。GFPT1 和该途径下游的其他基因的突变导致先天性肌无力综合征(CMS),其特征是由于神经传递受损而导致肌肉无力易疲劳。由于 GFPT1 缺乏导致神经肌肉接头(NMJ)的确切病理机制尚未发现。我们面临的挑战之一是 Gfpt1-/- 敲除小鼠的生存能力。在这项研究中,我们使用 Cre/LoxP 技术生成了一种肌肉特异性 GFPT1 敲除小鼠模型,Gfpt1tm1d/tm1d,具有人类 CMS 表型的特征。我们的数据表明,肌肉来源的 GFPT1 在 NMJ 的发育、神经传递、骨骼肌完整性中起着关键作用,并强调仅骨骼肌缺乏就足以引起形态后突触 NMJ 变化,这些变化伴随着前突触改变,尽管神经元 GFPT1 表达得到保留。除了常规的形态学 NMJ 变化和易疲劳的肌肉无力外,Gfpt1tm1d/tm1d 小鼠还表现出进行性肌病表型,肌肉中存在管状聚集物,这是 GFPT1-CMS 表型的特征。我们进一步发现骨骼肌蛋白聚糖-1、法呢基转移酶/香叶基香叶基转移酶 1 亚基α和肌肉特异性激酶的上调,这些蛋白已知参与 NMJ 的分化和维持。Gfpt1tm1d/tm1d 模型允许进一步研究 GFPT1 下游基因和途径的病理生理后果,这些基因和途径可能涉及 NMJ 和肌肉靶标的糖基化或低聚糖基化错误。