Department of Genetics, Pitié-Salpêtrière Hospital, Assistance Publique-Hôpitaux de Paris (AP-HP), Sorbonne Université, Paris, France.
INSERM UMR1170 and.
Blood. 2018 Sep 20;132(12):1318-1331. doi: 10.1182/blood-2017-12-820308. Epub 2018 Jun 18.
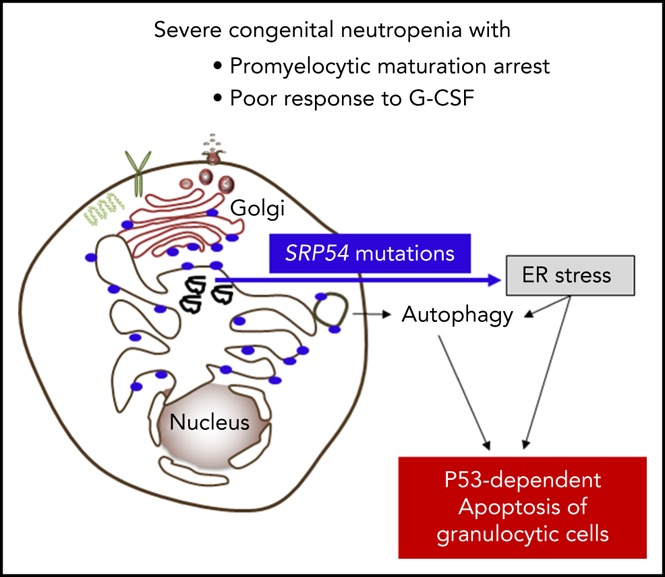
Congenital neutropenias (CNs) are rare heterogeneous genetic disorders, with about 25% of patients without known genetic defects. Using whole-exome sequencing, we identified a heterozygous mutation in the gene, encoding the signal recognition particle (SRP) 54 GTPase protein, in 3 sporadic cases and 1 autosomal dominant family. We subsequently sequenced the gene in 66 probands from the French CN registry. In total, we identified 23 mutated cases (16 sporadic, 7 familial) with 7 distinct germ line mutations including a recurrent in-frame deletion (Thr117del) in 14 cases. In nearly all patients, neutropenia was chronic and profound with promyelocytic maturation arrest, occurring within the first months of life, and required long-term granulocyte colony-stimulating factor therapy with a poor response. Neutropenia was sometimes associated with a severe neurodevelopmental delay (n = 5) and/or an exocrine pancreatic insufficiency requiring enzyme supplementation (n = 3). The SRP54 protein is a key component of the ribonucleoprotein complex that mediates the co-translational targeting of secretory and membrane proteins to the endoplasmic reticulum (ER). We showed that SRP54 was specifically upregulated during the in vitro granulocytic differentiation, and that mutations or knockdown led to a drastically reduced proliferation of granulocytic cells associated with an enhanced P53-dependent apoptosis. Bone marrow examination of -mutated patients revealed a major dysgranulopoiesis and features of cellular ER stress and autophagy that were confirmed using -mutated primary cells and knockdown cells. In conclusion, we characterized a pathological pathway, which represents the second most common cause of CN with maturation arrest in the French CN registry.
先天性中性粒细胞减少症(CNs)是一种罕见的异质性遗传疾病,约有 25%的患者没有已知的遗传缺陷。我们使用全外显子组测序,在 3 例散发性病例和 1 例常染色体显性家族中发现了 基因的杂合突变,该基因编码信号识别颗粒(SRP)54 GTP 酶蛋白。随后,我们在法国 CN 登记处的 66 名先证者中对 基因进行了测序。总共发现了 23 例突变病例(16 例散发性,7 例家族性),有 7 种不同的种系突变,包括 14 例中的重复框内缺失(Thr117del)。几乎所有患者的中性粒细胞减少症均为慢性和严重的,伴有早幼粒细胞成熟停滞,发生在生命的前几个月,需要长期接受粒细胞集落刺激因子治疗,但反应不佳。中性粒细胞减少症有时与严重的神经发育迟缓(n=5)和/或需要补充酶的外分泌胰腺功能不全(n=3)有关。SRP54 蛋白是介导分泌蛋白和膜蛋白共翻译靶向内质网(ER)的核糖核蛋白复合物的关键组成部分。我们表明,SRP54 在体外粒细胞分化过程中特异性上调,而 突变或敲低导致粒细胞细胞增殖明显减少,与增强的 P53 依赖性细胞凋亡有关。-突变患者的骨髓检查显示严重的粒系发育不良,并表现出细胞 ER 应激和自噬的特征,这些特征在使用 -突变的原代细胞和 敲低细胞中得到了证实。总之,我们描述了一种病理途径,该途径代表法国 CN 登记处以成熟停滞为特征的 CN 的第二大常见原因。