Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences (NDORMS), Botnar Research Centre, University of Oxford, Headington, Oxford, United Kingdom.
PLoS One. 2018 Jun 19;13(6):e0197883. doi: 10.1371/journal.pone.0197883. eCollection 2018.
All surgical meshes entering the U.S. market have been cleared for clinical use by the 510(k) process of the Food and Drug Administration (FDA), in which devices simply require proof of "substantial equivalence" to predicate devices, without the need for clinical trials. However, recalled meshes associated with adverse effects may, indirectly, continue to serve as predicates for new devices raising concerns over the safety of the 510(k) route.
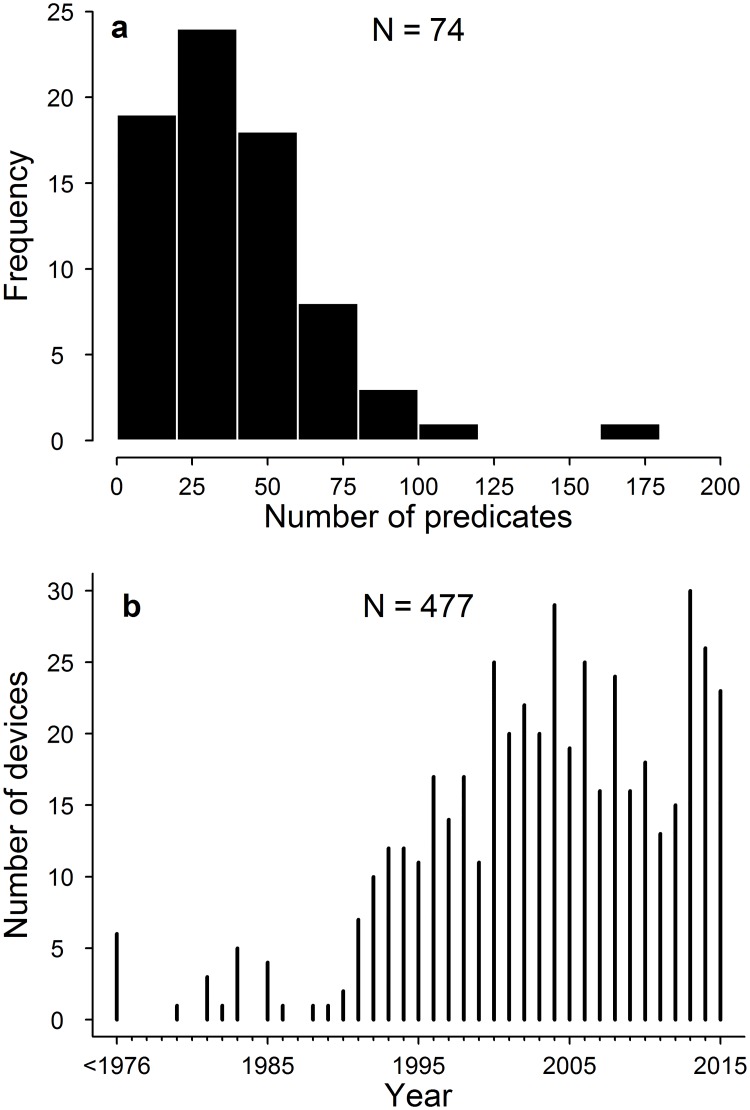
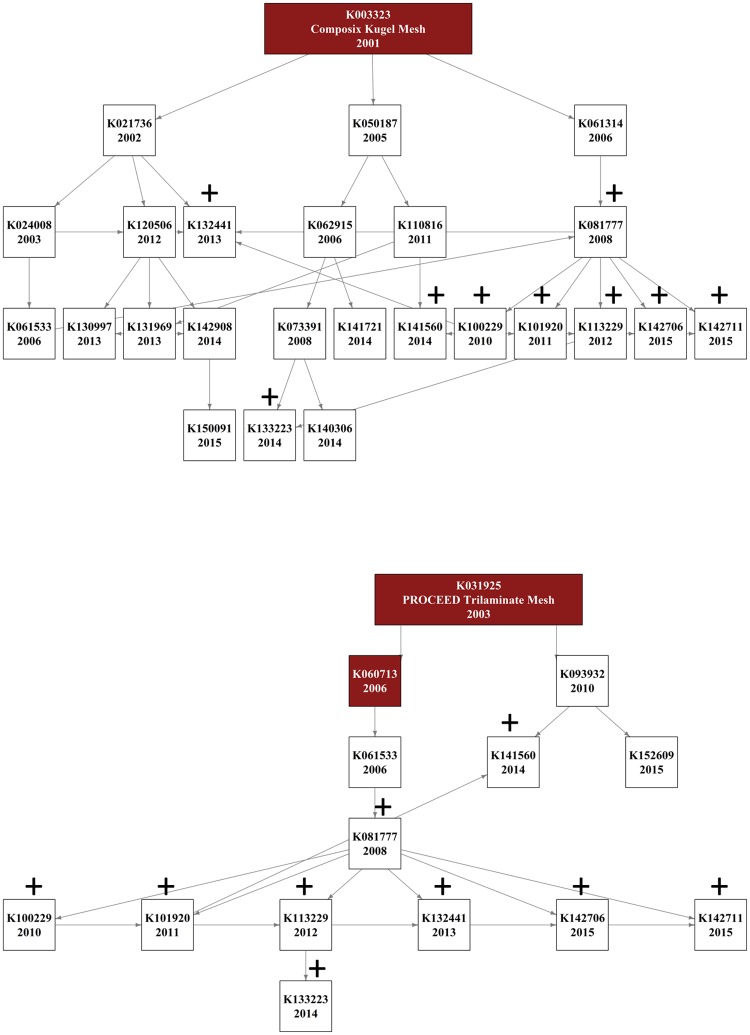
Here we assess the potential magnitude of this problem by determining the ancestral network of equivalence claims linking recently cleared surgical meshes. Using the FDA website we identified all surgical meshes cleared by the 510(k) route between January 2013 and December 2015 along with all listed predicates for these devices. Using a network approach, we trace the ancestry of predicates across multiple generations of equivalence claims and identify those meshes connected to devices that have since recalled from the market along with the reason for their recall.
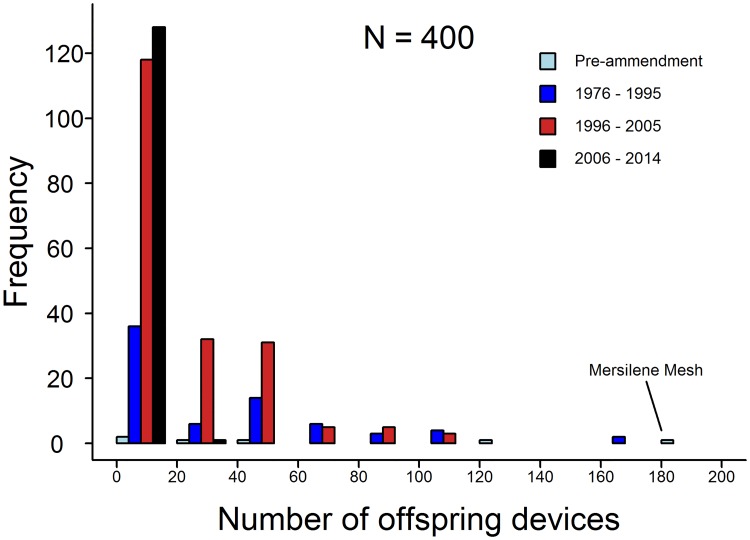
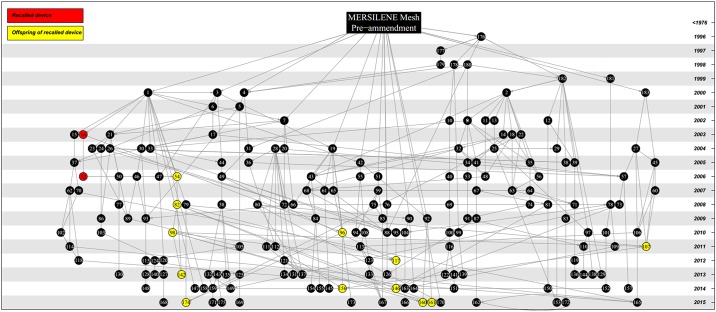
We find that the 77 surgical meshes cleared between 2013 and 2015 are based on 771 interconnected predicate claims of equivalence from 400 other devices. The vast majority of these devices (97%) are descended from only six surgical meshes that were present on the market prior to 1976. One of these ancestral meshes alone, provided the basis of 183 subsequent devices. Furthermore, we show that 16% of recently cleared devices are connected through equivalence claims to the 3 predicate meshes that have been recalled for design and material related flaws causing serious adverse events. Taken together, our results show that surgical meshes are connected through a tangled web of equivalency claims and many meshes recently cleared by the FDA have connections through chains of equivalency to devices which have been recalled from the market due to concerns over clinical safety. These findings raise concerns over the efficacy of the 510(k) route in ensuring patient safety.
所有进入美国市场的外科用网片都已通过食品和药物管理局(FDA)的 510(k)程序获得临床使用许可,在此程序中,器械只需证明与参比器械具有“实质等同性”,而无需进行临床试验。然而,与不良事件相关的召回网片可能会间接继续作为新器械的参比,这引发了人们对 510(k)途径安全性的担忧。
在这里,我们通过确定最近获得批准的外科用网片的等效性主张的祖先网络,评估这个问题的潜在程度。我们利用 FDA 网站,确定了 2013 年 1 月至 2015 年 12 月期间通过 510(k)途径获得批准的所有外科用网片,以及这些器械的所有上市参比。我们采用网络方法,追踪跨多代等效性主张的参比起源,并确定与已从市场召回的器械相连的网片,以及它们被召回的原因。
我们发现,2013 年至 2015 年间获得批准的 77 个外科用网片,是基于 400 个其他器械的 771 个相互关联的等效性主张。这些器械中的绝大多数(97%)源自于 1976 年之前就已上市的 6 个外科用网片。其中一个单独的祖先网片,为 183 个后续器械提供了基础。此外,我们还表明,最近获得批准的 16%的器械,通过等效性主张与 3 个因设计和材料缺陷导致严重不良事件而被召回的参比网片相连接。总的来说,我们的研究结果表明,外科用网片通过错综复杂的等效性主张网络连接在一起,许多最近获得 FDA 批准的网片通过等效性主张与因临床安全问题而从市场召回的器械相连接。这些发现引发了对 510(k)途径在确保患者安全方面的有效性的担忧。