Department of Molecular and Cell Biology, University of Leicester, Leicester, United Kingdom.
Department of Molecular and Cell Biology, University of Leicester, Leicester, United Kingdom; Leicester Institute of Structural and Chemical Biology, University of Leicester, Leicester, United Kingdom.
J Biol Chem. 2018 Aug 17;293(33):12820-12831. doi: 10.1074/jbc.RA118.003737. Epub 2018 Jul 11.
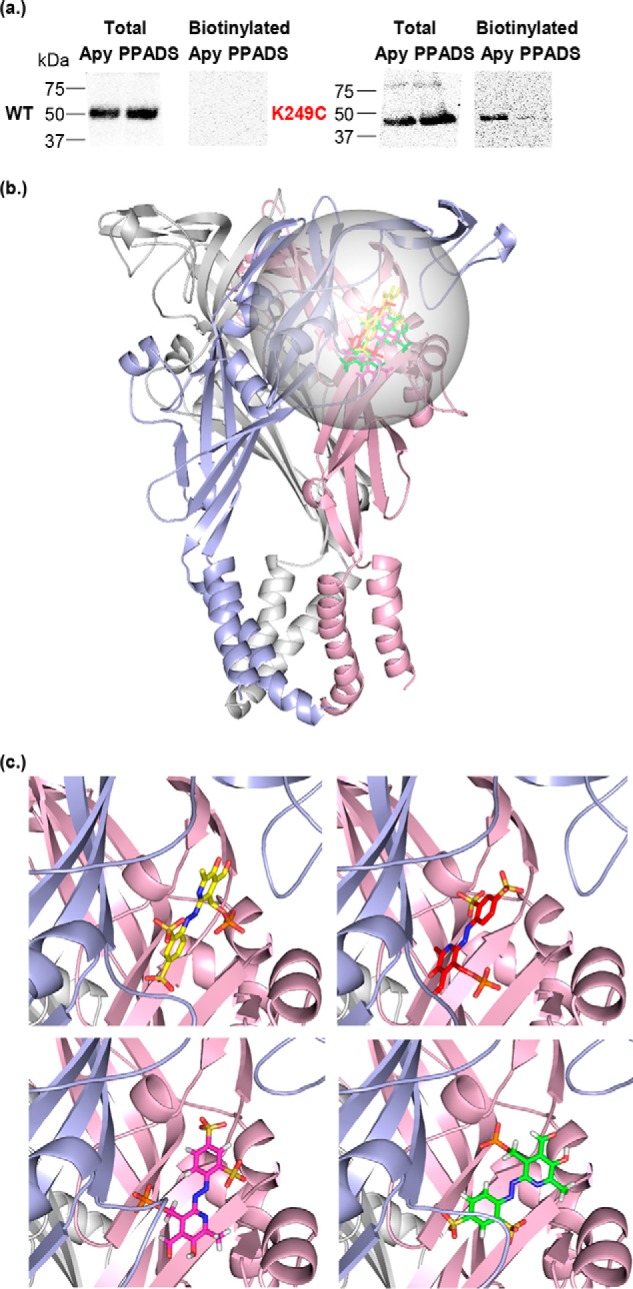
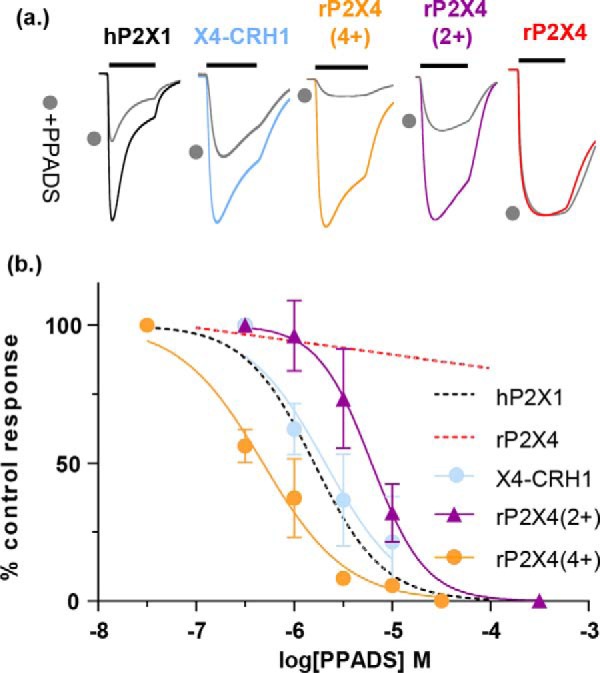
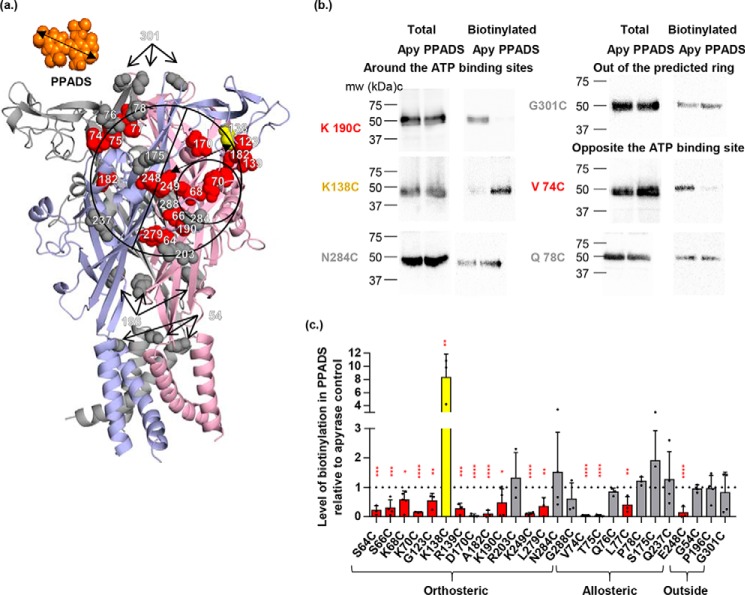
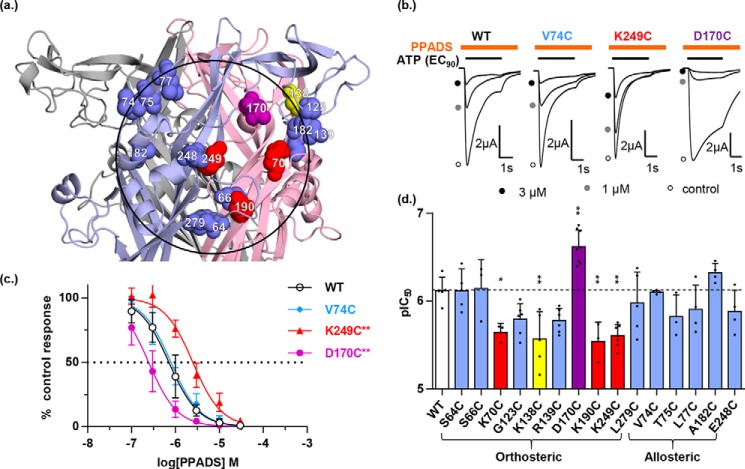
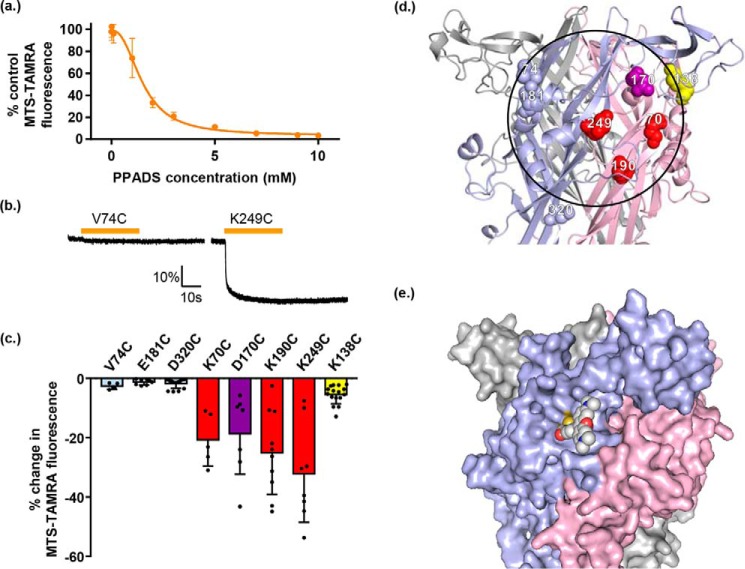
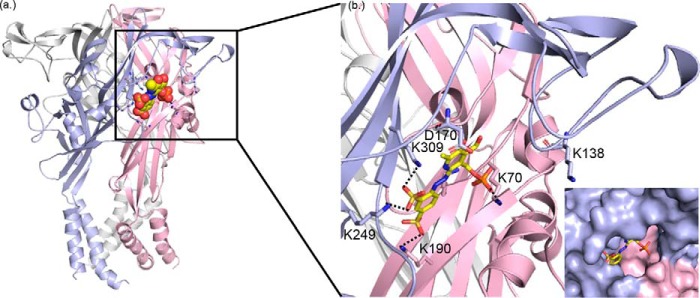
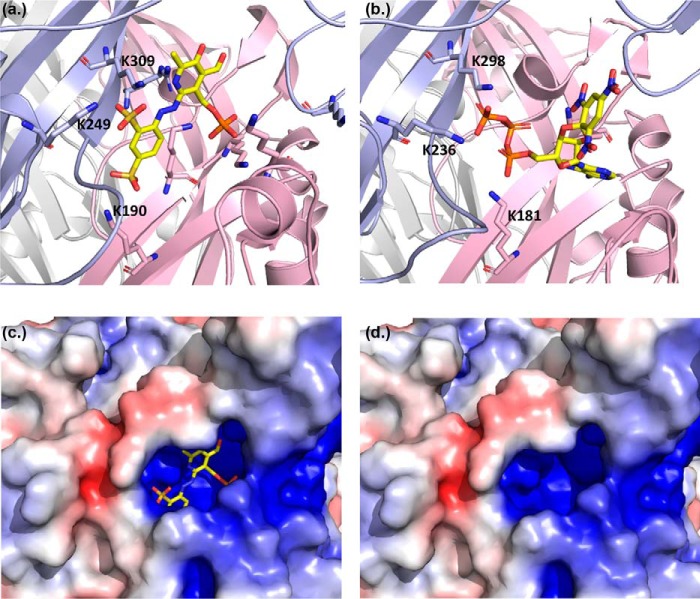
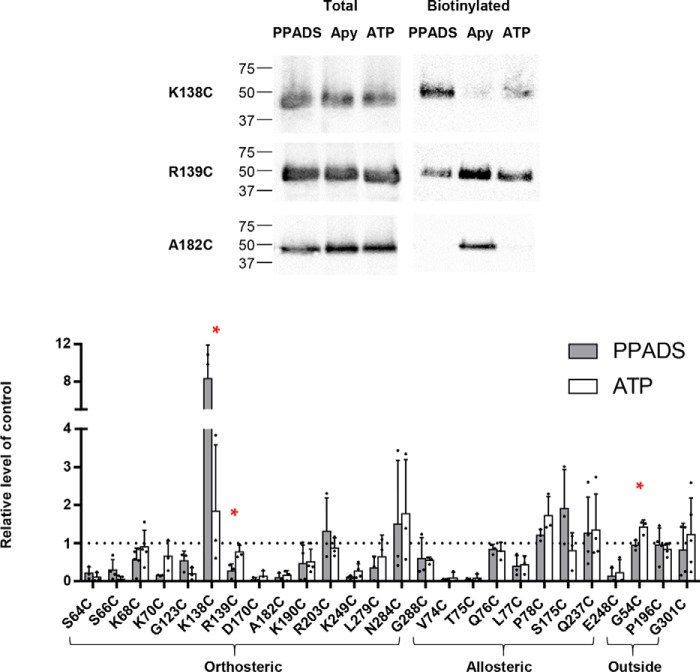
ATP is the native agonist for cell-surface ligand-gated P2X receptor (P2XR) cation channels. The seven mammalian subunits (P2X1-7) form homo- and heterotrimeric P2XRs having significant physiological and pathophysiological roles. Pyridoxalphosphate-6-azophenyl-2',4'-disulfonic acid (PPADS) is an effective antagonist at most mammalian P2XRs. Lys-249 in the extracellular domain of P2XR has previously been shown to contribute to PPADS action. To map this antagonist site, we generated human P2X1R cysteine substitutions within a circle centered at Lys-249 (with a radius of 13 Å equal to the length of PPADS). We hypothesized that cysteine substitutions of residues involved in PPADS binding would (i) reduce cysteine accessibility (measured by MTSEA-biotinylation), (ii) exhibit altered PPADS affinity, and (iii) quench the fluorescence of cysteine residues modified with MTS-TAMRA. Of the 26 residues tested, these criteria were met by only four (Lys-70, Asp-170, Lys-190, and Lys-249), defining the antagonist site, validating molecular docking results, and thereby providing the first experimentally supported model of PPADS binding. This binding site overlapped with the ATP-binding site, indicating that PPADS sterically blocks agonist access. Moreover, PPADS induced a conformational change at the cysteine-rich head (CRH) region adjacent to the orthosteric ATP-binding pocket. The importance of this movement was confirmed by demonstrating that substitution introducing positive charge present in the CRH of the hP2X1R causes PPADS sensitivity at the normally insensitive rat P2X4R. This study provides a template for developing P2XR subtype selectivity based on the differences among the mammalian subunits around the orthosteric P2XR-binding site and the CRH.
ATP 是细胞表面配体门控 P2X 受体 (P2XR) 阳离子通道的天然激动剂。七种哺乳动物亚基(P2X1-7)形成具有重要生理和病理生理作用的同型和异型三聚体 P2XR。吡哆醛-6-叠氮苯-2',4'-二磺酸(PPADS)是大多数哺乳动物 P2XR 的有效拮抗剂。先前已经表明,P2XR 细胞外结构域中的 Lys-249 残基有助于 PPADS 发挥作用。为了绘制这个拮抗剂结合位点,我们在以 Lys-249 为中心的圆内(半径为 13Å,等于 PPADS 的长度)生成了人类 P2X1R 半胱氨酸取代。我们假设参与 PPADS 结合的残基的半胱氨酸取代将 (i) 降低半胱氨酸可及性(通过 MTSEA-生物素化测量),(ii) 表现出改变的 PPADS 亲和力,和 (iii) 猝灭用 MTS-TAMRA 修饰的半胱氨酸残基的荧光。在测试的 26 个残基中,只有四个(Lys-70、Asp-170、Lys-190 和 Lys-249)满足这些标准,定义了拮抗剂结合位点,验证了分子对接结果,从而提供了第一个实验支持的 PPADS 结合模型。该结合位点与 ATP 结合位点重叠,表明 PPADS 空间上阻止激动剂进入。此外,PPADS 在紧邻正构 ATP 结合口袋的富含半胱氨酸的头部(CRH)区域诱导构象变化。通过证明在 hP2X1R 的 CRH 中引入正电荷的取代会导致通常对 rat P2X4R 不敏感的 PPADS 敏感性,证实了这种运动的重要性。这项研究为基于正构 P2XR 结合位点和 CRH 周围哺乳动物亚基之间的差异开发 P2XR 亚型选择性提供了模板。