Lu Jingru, Zhao Xiangzhong, Paiardini Alessandro, Lang Yanhua, Bottillo Irene, Shao Leping
Department of Nephrology, The Affiliated Hospital of Qingdao University, 16 Jiangsu Road, Qingdao, 266003, China.
Central Laboratory, The Affiliated Hospital of Qingdao University, 1677 Wutaishan Road, Qingdao, 266555, China.
BMC Nephrol. 2018 Jul 13;19(1):181. doi: 10.1186/s12882-018-0979-1.
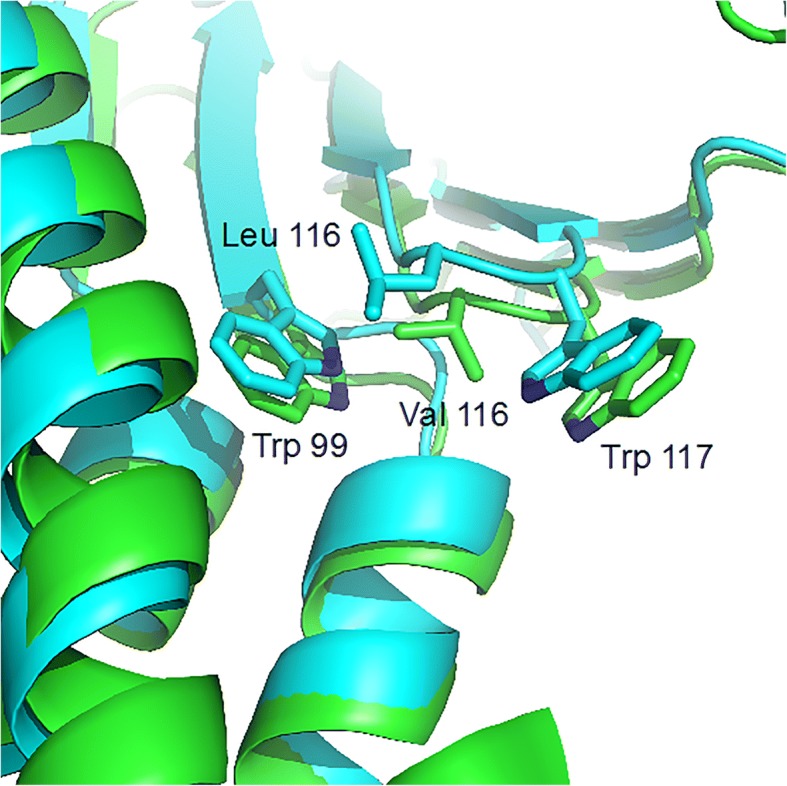
Sixty mutations of claudin 16 coding gene have been reported in familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC) patients. Recent investigations revealed that a highly conserved glycine-leucine-tryptophan (G-L-W) motif in the first extracellular segment (ESC1) of claudin 16 might be essential for stabilization of the appropriately folded ECS1 structure and conservation of normal claudin 16 function. However, neither missense nor nonsense mutation has ever been described in this motif. Our study aimed at identifying mutations in a Chinese patient with FHHNC and exploring the association between genotype and phenotype.
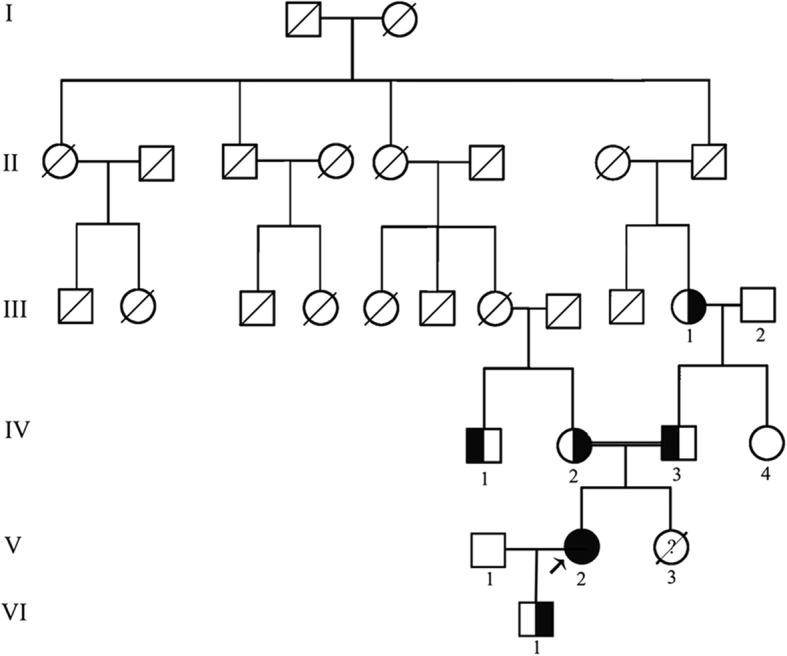
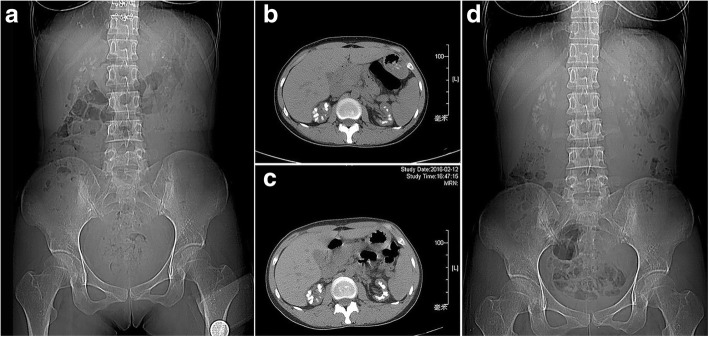
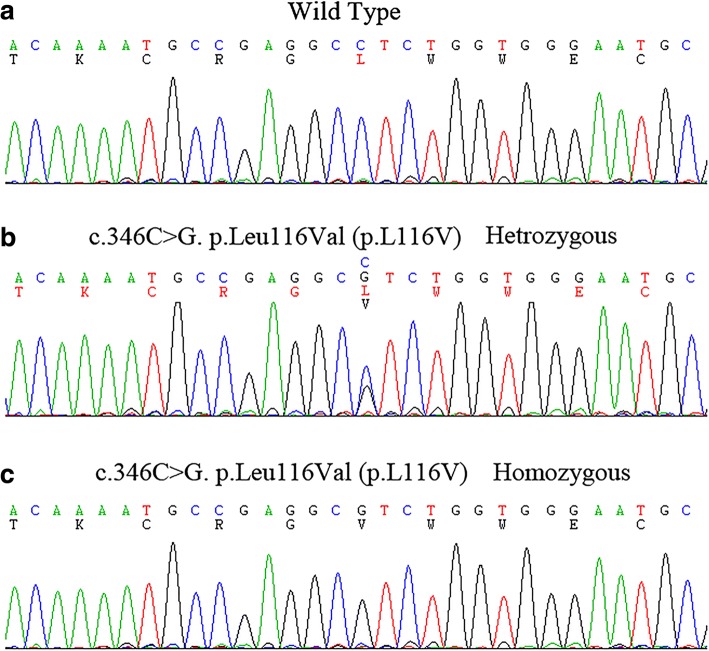
A 33-year-old female presented with 4 years history of recurrent acute pyelonephritis without other notable past medical history. Her healthy parents, who aged 56 and 53 respectively, were second cousins, and her only sibling died from renal failure without definite cause at age 25. Renal ultrasound imaging demonstrated atrophic kidneys and bilateral nephrocalcinosis. The laboratory workup revealed impaired renal function (Stage CKD IV), hypocalcemia and mild hypomagnesemia, accompanied with marked renal loss of magnesium and hypercalciuria. During the follow-up, treatment with calcitriol and calcium but not with magnesium was difficult to achieve normal serum calcium levels, whereas her serum magnesium concentration fluctuated within normal ranges. In the end, the patient unavoidably reached ESRD at 36 years old. The clinical features and family history suggested the diagnosis of FHHNC. To make a definite diagnosis, we use whole-exome sequencing to identify the disease-causing mutations and Sanger sequencing to confirm the mutation co-segregation in the family. As a result, a novel homozygous mutation (c.346C > G, p.Leu116Val) in G-L-W motif of claudin 16 was identified. Her parents, grandmother and one of her cousins carried heterozygous p.Leu116Val, whereas 200 unrelated controls did not carry this mutation.
We described a delayed diagnosis patient with FHHNC in the Chinese population and identified a novel missense mutation in the highly conserved G-L-W motif of claudin 16 for the first time. According to the reported data and the information deduced from 3D modeling, we speculate that this mutation probably reserve partial residual function which might be related to the slight phenotype of the patient.
在家族性低镁血症伴高钙尿症和肾钙质沉着症(FHHNC)患者中,已报道了60种claudin 16编码基因突变。最近的研究表明,claudin 16第一个细胞外结构域(ESC1)中高度保守的甘氨酸 - 亮氨酸 - 色氨酸(G-L-W)基序可能对稳定正确折叠的ECS1结构和维持正常的claudin 16功能至关重要。然而,该基序中从未描述过错义突变或无义突变。我们的研究旨在鉴定一名中国FHHNC患者的突变,并探讨基因型与表型之间的关联。
一名33岁女性有4年复发性急性肾盂肾炎病史,无其他明显既往病史。她健康的父母分别为56岁和53岁,是二级表亲,她唯一的兄弟姐妹在25岁时死于不明原因的肾衰竭。肾脏超声成像显示肾脏萎缩和双侧肾钙质沉着症。实验室检查显示肾功能受损(慢性肾脏病IV期)、低钙血症和轻度低镁血症,伴有明显的肾脏镁丢失和高钙尿症。在随访期间,使用骨化三醇和钙治疗但不使用镁治疗难以使血清钙水平恢复正常,而她的血清镁浓度在正常范围内波动。最终,该患者在36岁时不可避免地发展为终末期肾病(ESRD)。临床特征和家族史提示FHHNC的诊断。为明确诊断,我们使用全外显子测序来鉴定致病突变,并使用桑格测序来确认家族中的突变共分离情况。结果,在claudin 16的G-L-W基序中鉴定出一个新的纯合突变(c.346C>G,p.Leu116Val)。她的父母、祖母和她的一个表亲携带杂合的p.Leu116Val,而200名无关对照未携带此突变。
我们描述了一名中国人群中诊断延迟的FHHNC患者,并首次在claudin 16高度保守的G-L-W基序中鉴定出一个新的错义突变。根据已报道的数据和从三维建模推导的信息,我们推测该突变可能保留了部分残余功能,这可能与患者的轻微表型有关。