Respiratory Translational Research Group, Department of Laboratory Medicine, College of Health and Medicine, University of Tasmania, Launceston, Tasmania 7248, Australia.
Woolcock Emphysema Centre, Woolcock Institute of Medical Research, The University of Sydney, Sydney, NSW 2037, Australia.
Clin Sci (Lond). 2018 Jul 31;132(14):1615-1627. doi: 10.1042/CS20180398.
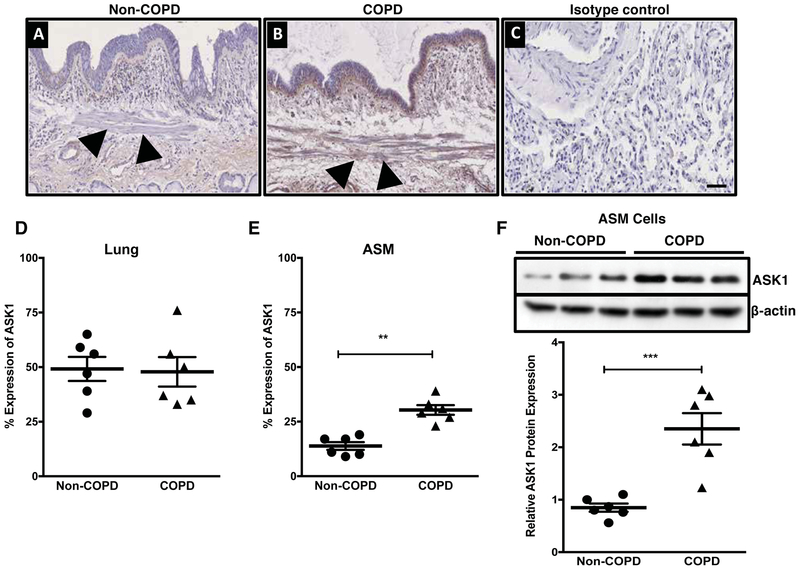
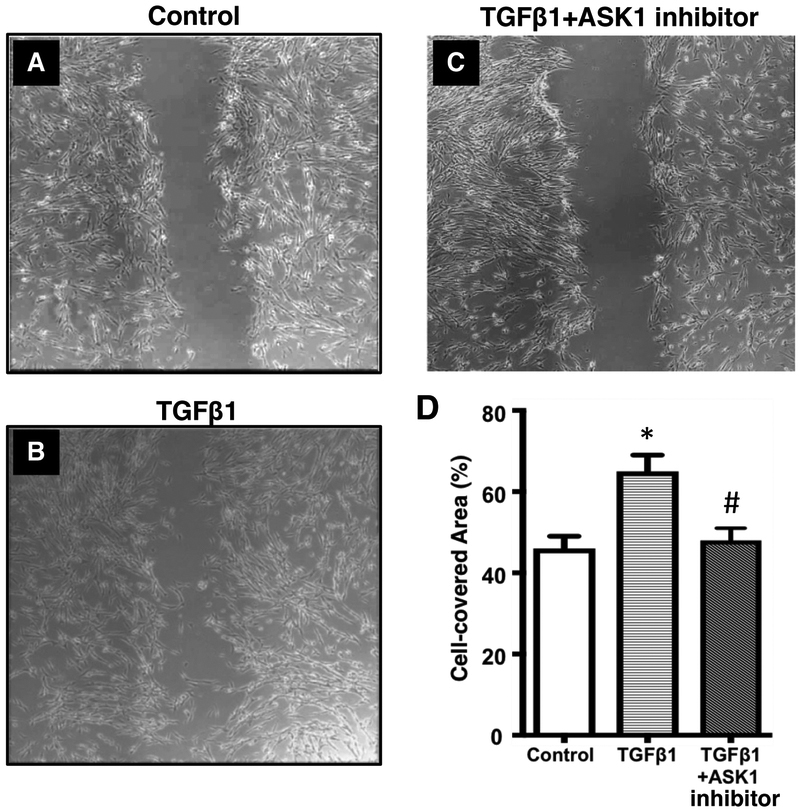
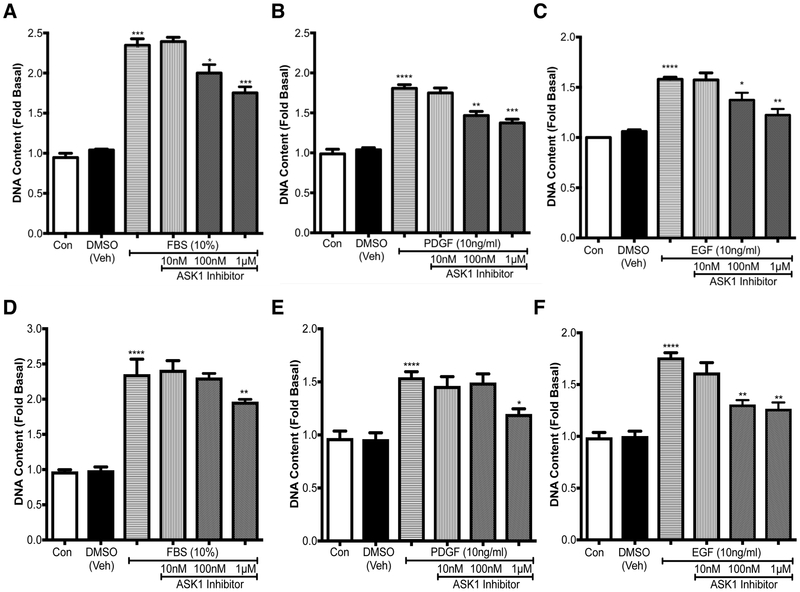
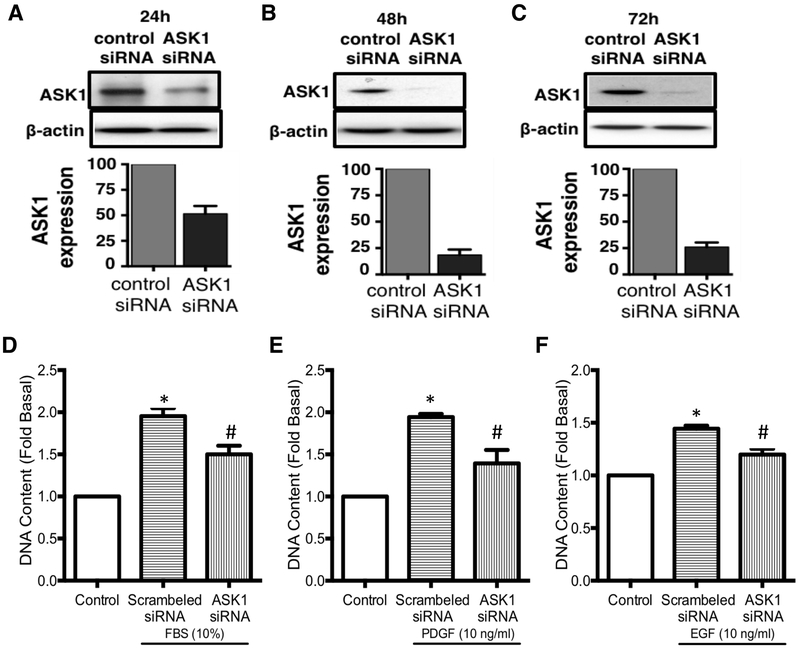
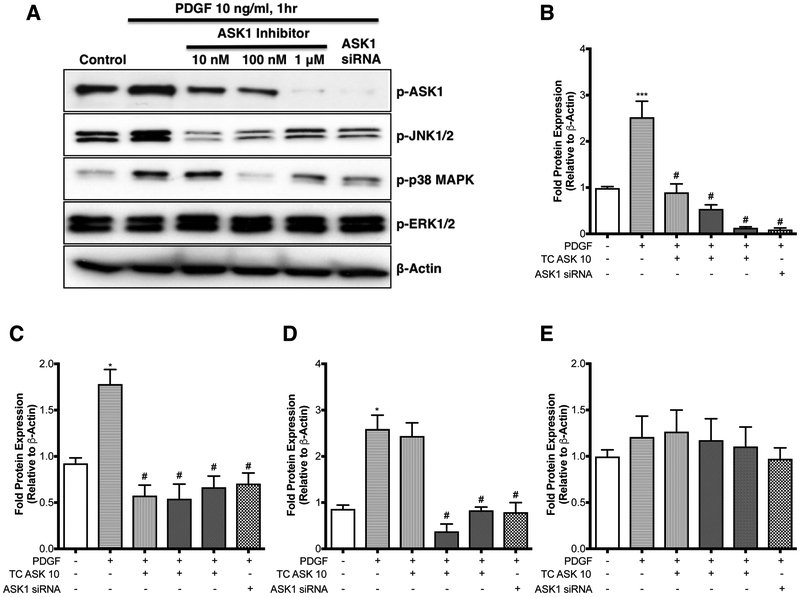
Increased airway smooth muscle (ASM) mass is observed in chronic obstructive pulmonary disease (COPD), which is correlated with disease severity and negatively affects lung function in these patients. Thus, there is clear unmet clinical need for finding new therapies which can target airway remodeling and disease progression in COPD. Apoptosis signal-regulating kinase 1 (ASK1) is a ubiquitously expressed mitogen-activated protein kinase (MAPK) kinase kinase (MAP3K) activated by various stress stimuli, including reactive oxygen species (ROS), tumor necrosis factor (TNF)-α, and lipopolysaccharide (LPS) and is known to regulate cell proliferation. ASM cells from COPD patients are hyperproliferative to mitogens However, the role of ASK1 in ASM growth is not established. Here, we aim to determine the effects of ASK1 inhibition on ASM growth and pro-mitogenic signaling using ASM cells from COPD patients. We found greater expression of ASK1 in ASM bundles of COPD lung when compared with non-COPD. Pre-treatment of ASM cells with highly selective ASK1 inhibitor, TC ASK 10 resulted in a dose-dependent reduction in mitogen (FBS, PDGF, and EGF; 72 h)-induced ASM growth as measured by CyQUANT assay. Further, molecular targetting of ASK1 using siRNA in ASM cells prevented mitogen-induced cell growth. In addition, to anti-mitogenic potential, ASK1 inhibitor also prevented TGFβ1-induced migration of ASM cells Immunoblotting revealed that anti-mitogenic effects are mediated by C-Jun N-terminal kinase (JNK) and p38MAP kinase-signaling pathways as evident by reduced phosphorylation of downstream effectors JNK1/2 and p38MAP kinases, respectively, with no effect on extracellular signal-regulated kinase (ERK) 1/2 (ERK1/2). Collectively, these findings establish the anti-mitogenic effect of ASK1 inhibition and identify a novel pathway that can be targetted to reduce or prevent excessive ASM mass in COPD.
在慢性阻塞性肺疾病(COPD)中观察到气道平滑肌(ASM)质量增加,这与疾病严重程度相关,并对这些患者的肺功能产生负面影响。因此,对于寻找新的治疗方法来靶向 COPD 中的气道重塑和疾病进展,存在明显的未满足的临床需求。凋亡信号调节激酶 1(ASK1)是一种广泛表达的丝裂原激活蛋白激酶(MAPK)激酶激酶(MAP3K),可被各种应激刺激物激活,包括活性氧(ROS)、肿瘤坏死因子(TNF)-α和脂多糖(LPS),并已知其可调节细胞增殖。COPD 患者的 ASM 细胞对有丝分裂原呈过度增殖状态。然而,ASK1 在 ASM 生长中的作用尚未确定。在这里,我们旨在使用来自 COPD 患者的 ASM 细胞来确定 ASK1 抑制对 ASM 生长和促有丝分裂信号的影响。我们发现,与非 COPD 相比,COPD 肺中的 ASM 束中 ASK1 的表达更高。用高选择性 ASK1 抑制剂 TC ASK 10 预处理 ASM 细胞可剂量依赖性地减少有丝分裂原(FBS、PDGF 和 EGF;72 h)诱导的 ASM 生长,如 CyQUANT 测定法所测量的。此外,在 ASM 细胞中使用 siRNA 对 ASK1 进行分子靶向处理可防止有丝分裂原诱导的细胞生长。此外,ASK1 抑制剂除了具有抗有丝分裂作用外,还可防止 TGFβ1 诱导的 ASM 细胞迁移。免疫印迹显示,抗有丝分裂作用是通过减少下游效应子 JNK1/2 和 p38MAP 激酶的磷酸化来介导的,分别是 JNK 和 p38MAP 激酶信号通路,对细胞外信号调节激酶(ERK)1/2(ERK1/2)没有影响。总的来说,这些发现确立了 ASK1 抑制的抗有丝分裂作用,并确定了一种新的途径,可以靶向该途径来减少或预防 COPD 中的过度 ASM 质量。