Watanabe Takanori, Yamazaki Sumire, Maita Chinatsu, Matushita Mizue, Matsuo Junji, Okubo Torahiko, Yamaguchi Hiroyuki
Department of Medical Laboratory Science, Faculty of Health Sciences, Hokkaido University, Sapporo, Japan.
Evol Bioinform Online. 2018 Jul 17;14:1176934318788337. doi: 10.1177/1176934318788337. eCollection 2018.
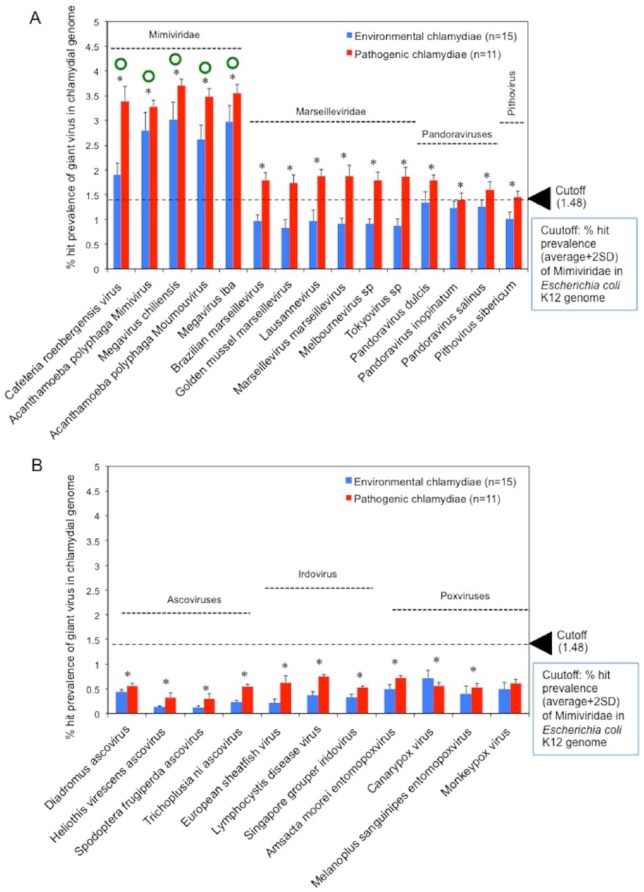
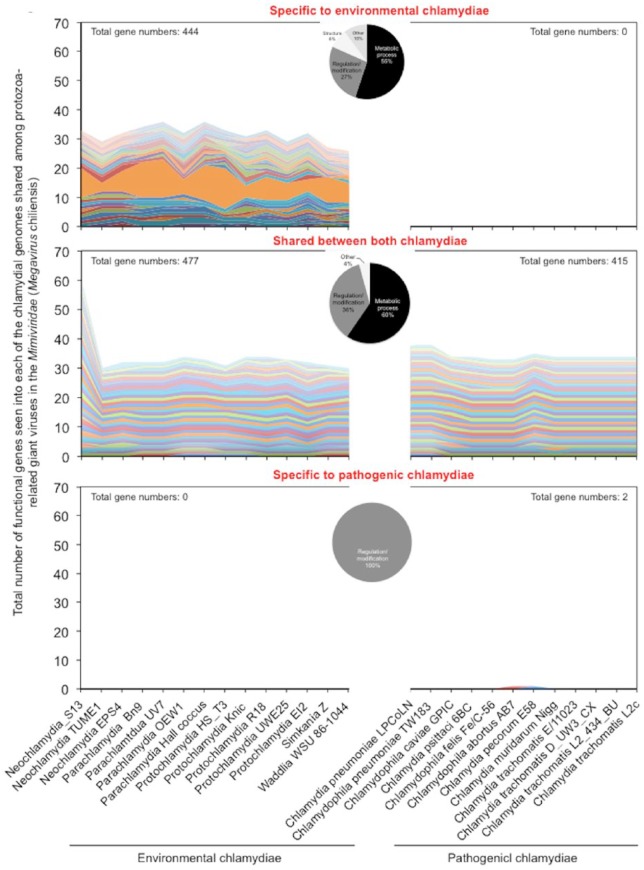
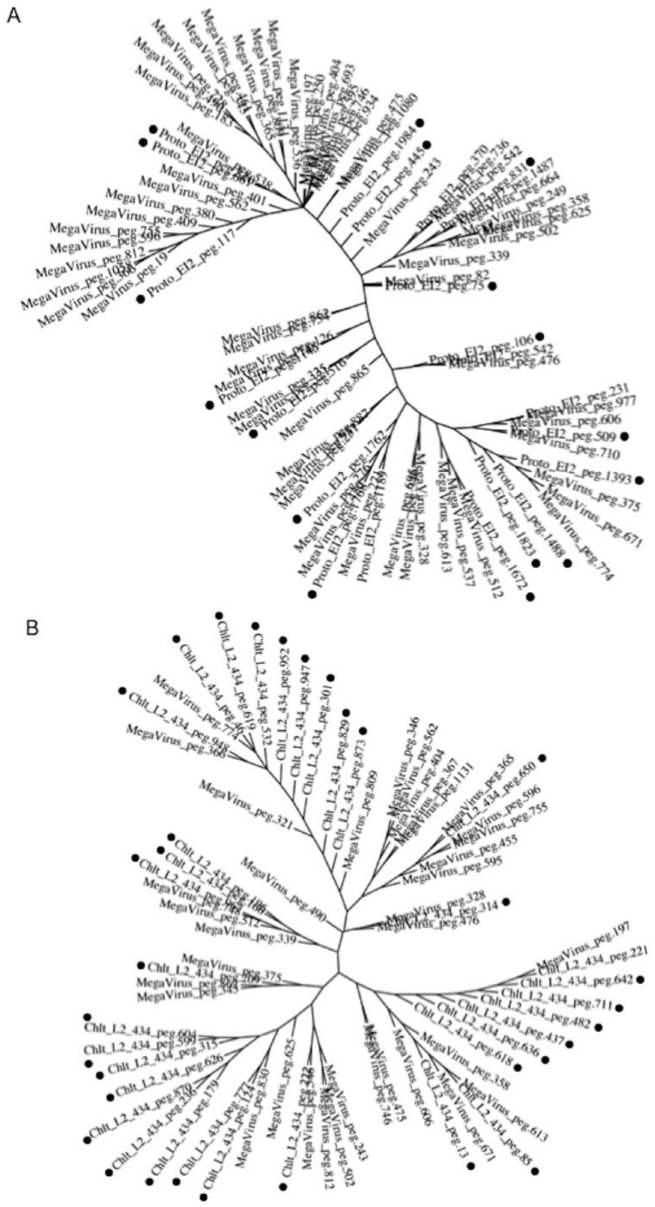
Obligate intracellular chlamydiae diverged into pathogenic and environmental chlamydiae 0.7-1.4 billion years ago. While pathogenic chlamydiae have adapted to a wide range of vertebrates, environmental chlamydiae inhabit unicellular amoebae, the free-living . However, how and why this divergence occurred remains unclear. Meanwhile, giant viruses consisting of protozoa-related and protozoa-unrelated viruses have been discovered, with the former group being suggested to have more influenced environmental chlamydiae during their evolution while cohabiting host amoebae. Against this background, we attempted to visualize genes of giant viruses in chlamydial genomes by bioinformatic analysis mainly with comparative genome and phylogenic analysis, seeking genes present in chlamydiae that are specifically shared with protozoa-related giant viruses. As a result, in contrast to protozoa-unrelated giant viruses, the genes of protozoa-related giant viruses were significantly shared in both the chlamydia genomes depending on the giant virus type. In particular, the prevalence of genes among the protozoa-related giant virus genes in chlamydial genomes was significantly high. Meanwhile, the prevalence of protozoa-related giant virus genes in pathogenic chlamydia genomes was consistently higher than those of environmental chlamydiae; the actual number of sequences similar to giant virus was also significantly predominant compared with those in the environmental chlamydial genomes. Among them, the most prevalent of giant virus was in the case of chlamydiae with ; total of 1338 genes of the chlamydiae were found to be shared with the virus (444 genes specific to environmental chlamydiae, 892 genes shared between both chlamydiae, only two genes in the pathogenic chlamydiae). Phylogenic analysis with most prevalent sets ( and EI2 or L2 434Bu) showed the presence of orthologs between these with several clustered. In addition, Pearson's single regression analysis revealed that almost the prevalence of the genes from the giant viruses in chlamydial genomes was negatively and specifically correlated with the number of chlamydial open reading frames (ORFs). Thus, these results indicated the trace of lateral gene transfer between protozoa-related giant viruses of family and chlamydiae. This is the first demonstration of a putative linkage between chlamydiae and the giant viruses, providing us with a hint to understand chlamydial evolution.
专性细胞内衣原体在 7 亿至 14 亿年前分化为致病性衣原体和环境衣原体。致病性衣原体已适应多种脊椎动物,而环境衣原体则寄生于单细胞变形虫等自由生活的生物中。然而,这种分化是如何以及为何发生的仍不清楚。与此同时,已发现由与原生动物相关和与原生动物无关的病毒组成的巨型病毒,有人认为前者在与宿主变形虫共同生活的进化过程中对环境衣原体的影响更大。在此背景下,我们主要通过比较基因组和系统发育分析等生物信息学分析方法,试图在衣原体基因组中可视化巨型病毒的基因,寻找衣原体中与原生动物相关巨型病毒特有的共享基因。结果发现,与与原生动物无关的巨型病毒不同,与原生动物相关巨型病毒的基因根据巨型病毒类型在两种衣原体基因组中均有显著共享。特别是,与原生动物相关巨型病毒基因在衣原体基因组中的流行率显著较高。同时,致病性衣原体基因组中与原生动物相关巨型病毒基因的流行率始终高于环境衣原体;与巨型病毒相似的序列实际数量与环境衣原体基因组相比也显著占优。其中,巨型病毒最普遍的情况是在具有[具体衣原体类型未明确给出]的衣原体中;发现该衣原体共有 1338 个基因与该病毒共享(444 个基因是环境衣原体特有的,892 个基因是两种衣原体共有的,致病性衣原体中只有两个基因)。对最普遍的基因集([具体基因集未明确给出]和 EI2 或 L2 434Bu)进行系统发育分析表明,这些基因之间存在直系同源物且有几个聚类。此外,皮尔逊单回归分析显示,衣原体基因组中来自巨型病毒的基因流行率几乎与衣原体开放阅读框(ORF)的数量呈负相关且具有特异性。因此,这些结果表明在[具体病毒科未明确给出]科与原生动物相关的巨型病毒和衣原体之间存在横向基因转移的痕迹。这是首次证明衣原体与巨型病毒之间存在推定联系,为我们理解衣原体进化提供了线索。