Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO 80309, United States of America.
PLoS One. 2018 Sep 6;13(9):e0202764. doi: 10.1371/journal.pone.0202764. eCollection 2018.
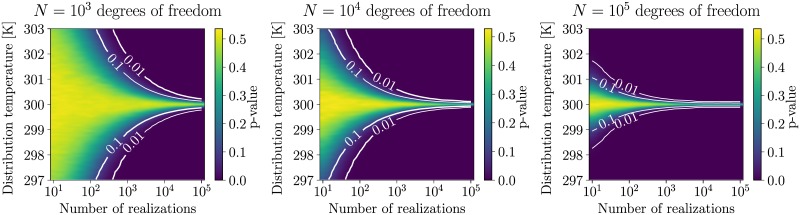
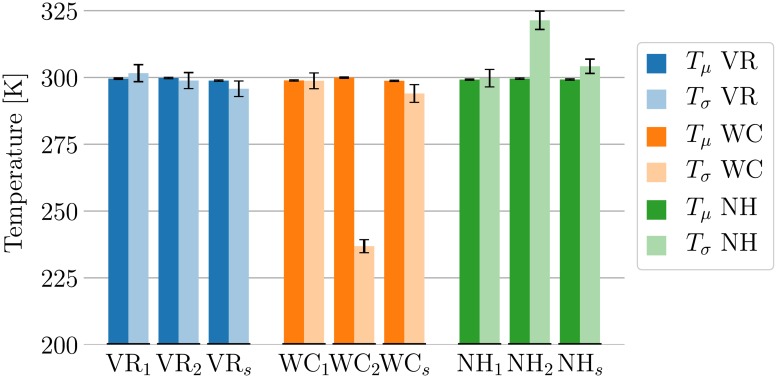
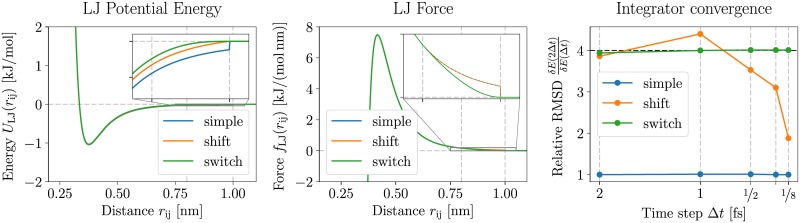
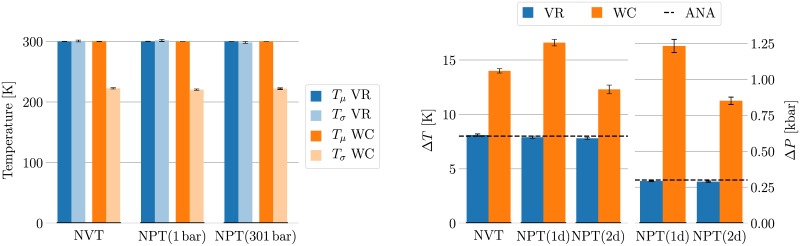
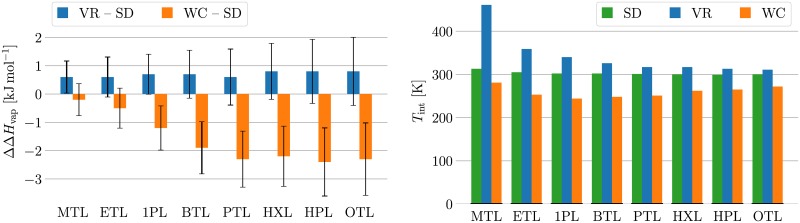
Advances in recent years have made molecular dynamics (MD) and Monte Carlo (MC) simulations powerful tools in molecular-level research, allowing the prediction of experimental observables in the study of systems such as proteins, membranes, and polymeric materials. However, the quality of any prediction based on molecular dynamics results will strongly depend on the validity of underlying physical assumptions. Unphysical behavior of simulations can have significant influence on the results and reproducibility of these simulations, such as folding of proteins and DNA or properties of lipid bilayers determined by cutoff treatment, dynamics of peptides and polymers affected by the choice of thermostat, or liquid properties depending on the simulation time step. Motivated by such examples, we propose a two-fold approach to increase the robustness of molecular simulations. The first part of this approach involves tests which can be performed by the users of MD programs on their respective systems and setups. We present a number of tests of different complexity, ranging from simple post-processing analysis to more involved tests requiring additional simulations. These tests are shown to significantly increase the reliability of MD simulations by catching a number of common simulation errors violating physical assumptions, such as non-conservative integrators, deviations from the Boltzmann ensemble, and lack of ergodicity between degrees of freedom. To make the usage as easy as possible, we have developed an open-source and platform-independent Python library (https://physical-validation.readthedocs.io) implementing these tests. The second part of the approach involves testing for code correctness. While unphysical behavior can be due to poor or incompatible choices of parameters by the user, it can just as well originate in coding errors within the program. We therefore propose to include physical validation tests in the code-checking mechanism of MD software packages. We have implemented such a validation for the GROMACS software package, ensuring that every major release passes a number of physical sanity checks performed on selected representative systems before shipping. It is, to our knowledge, the first major molecular mechanics software package to run such validation routinely. The tests are, as the rest of the package, open source software, and can be adapted for other software packages.
近年来的进展使得分子动力学(MD)和蒙特卡罗(MC)模拟成为分子水平研究的有力工具,使人们能够预测蛋白质、膜和聚合材料等系统的实验可观测结果。然而,任何基于分子动力学结果的预测的质量都将强烈取决于基础物理假设的有效性。模拟的非物理行为会对这些模拟的结果和可重复性产生重大影响,例如蛋白质和 DNA 的折叠,或截止处理确定的脂质双层的性质,选择恒温器会影响肽和聚合物的动力学,或取决于模拟时间步长的液体性质。受此类示例的启发,我们提出了一种提高分子模拟稳健性的双重方法。该方法的第一部分涉及 MD 程序的用户可以在其各自的系统和设置上执行的测试。我们提出了一系列不同复杂度的测试,从简单的后处理分析到更复杂的需要额外模拟的测试。这些测试通过捕获违反物理假设的许多常见模拟错误,例如非守恒积分器、偏离玻尔兹曼系综以及自由度之间缺乏遍历性,显著提高了 MD 模拟的可靠性。为了使使用尽可能简单,我们开发了一个开源且与平台无关的 Python 库(https://physical-validation.readthedocs.io)来实现这些测试。该方法的第二部分涉及代码正确性测试。虽然非物理行为可能是由于用户选择的参数不合适或不兼容造成的,但它也可能源自程序中的编码错误。因此,我们建议在 MD 软件包的代码检查机制中包含物理验证测试。我们已经为 GROMACS 软件包实现了这种验证,确保在发布之前,每个主要版本都会在选定的代表性系统上通过一些物理合理性检查。据我们所知,这是第一个常规运行此类验证的主要分子力学软件包。这些测试与包的其余部分一样是开源软件,可以适应其他软件包。