CERVO Research Center, Institut universitaire en santé mentale de Québec, Quebec City, QC, G1J 2G3, Canada.
Department of Biomedicine, University Hospital Basel, Basel, Switzerland.
Sci Rep. 2018 Sep 14;8(1):13804. doi: 10.1038/s41598-018-31772-0.
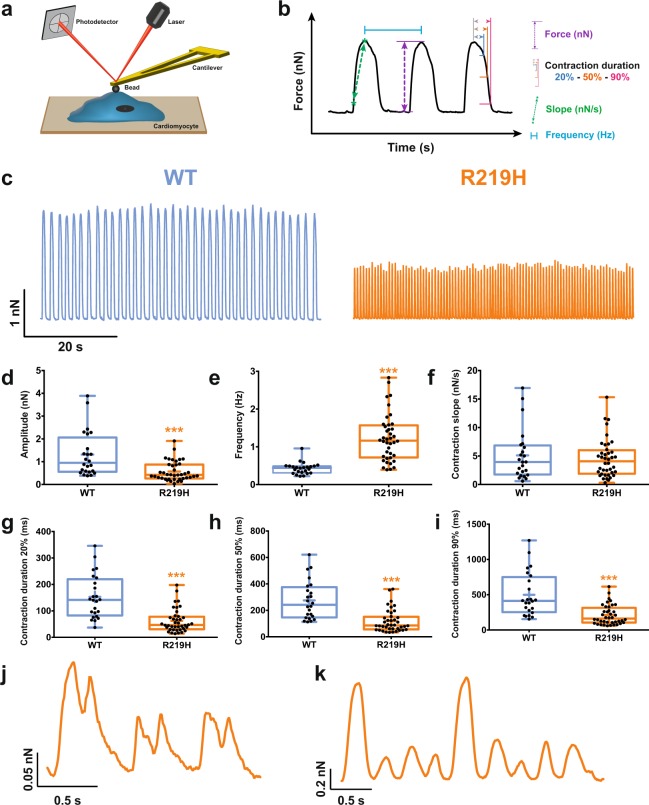
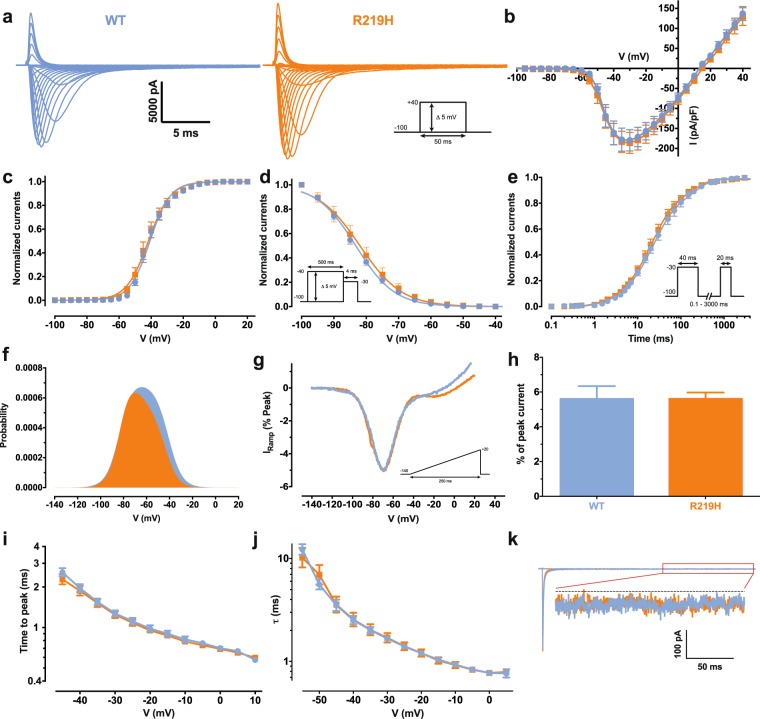
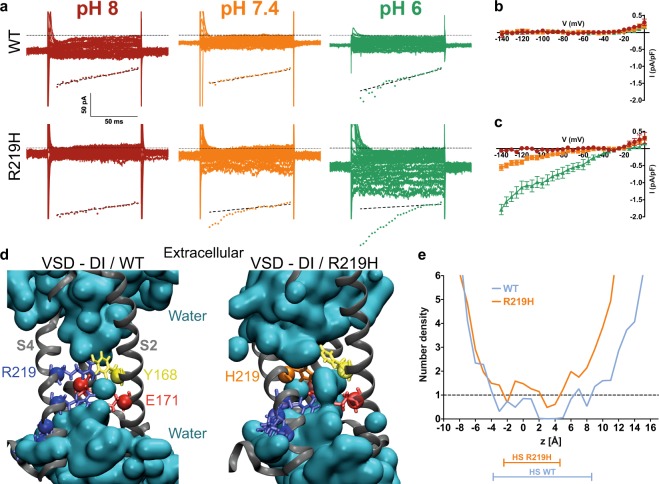
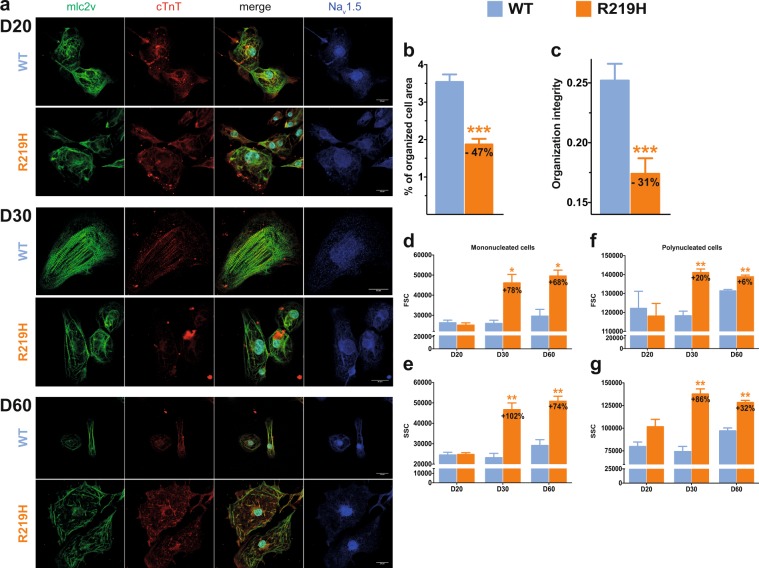
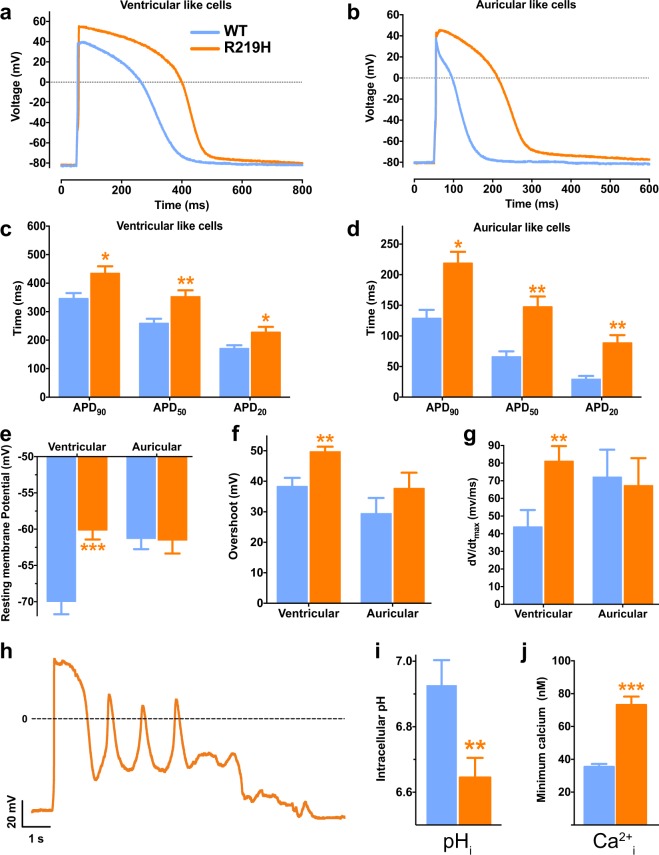
Dilated cardiomyopathy (DCM) is a structural heart disease that causes dilatation of cardiac chambers and impairs cardiac contractility. The SCN5A gene encodes Na1.5, the predominant cardiac sodium channel alpha subunit. SCN5A mutations have been identified in patients with arrhythmic disorders associated with DCM. The characterization of Na1.5 mutations located in the voltage sensor domain (VSD) and associated with DCM revealed divergent biophysical defects that do not fully explain the pathologies observed in these patients. The purpose of this study was to characterize the pathological consequences of a gating pore in the heart arising from the Na1.5/R219H mutation in a patient with complex cardiac arrhythmias and DCM. We report its properties using cardiomyocytes derived from patient-specific human induced pluripotent stem cells. We showed that this mutation generates a proton leak (called gating pore current). We also described disrupted ionic homeostasis, altered cellular morphology, electrical properties, and contractile function, most probably linked to the proton leak. We thus propose a novel link between SCN5A mutation and the complex pathogenesis of cardiac arrhythmias and DCM. Furthermore, we suggest that leaky channels would constitute a common pathological mechanism underlying several neuronal, neuromuscular, and cardiac pathologies.
扩张型心肌病(DCM)是一种结构性心脏病,可导致心脏腔室扩张并损害心脏收缩力。SCN5A 基因编码 Na1.5,即主要的心脏钠离子通道α亚基。心律失常疾病与 DCM 相关的患者中已发现 SCN5A 突变。位于电压传感器域(VSD)并与 DCM 相关的 Na1.5 突变的特征显示出不同的生物物理缺陷,这些缺陷不能完全解释在这些患者中观察到的病理学。本研究的目的是描述源自具有复杂心律失常和 DCM 的患者的 Na1.5/R219H 突变引起的心脏门控孔的病理后果。我们使用源自患者特异性人诱导多能干细胞的心肌细胞来报告其特性。我们表明该突变会产生质子泄漏(称为门控孔电流)。我们还描述了离子动态平衡、细胞形态、电特性和收缩功能的改变,这些改变很可能与质子泄漏有关。因此,我们提出了 SCN5A 突变与心律失常和 DCM 的复杂发病机制之间的新联系。此外,我们认为漏孔通道将构成几种神经元、神经肌肉和心脏病理学的常见病理机制。