Department of Medicine III, Institute for Cardiomyopathies Heidelberg (ICH), University of Heidelberg, 69120 Heidelberg, Germany.
DZHK (German Centre for Cardiovascular Research), Heidelberg-Mannheim, 17475 Greifswald, Germany.
Int J Mol Sci. 2021 Nov 30;22(23):12990. doi: 10.3390/ijms222312990.
Familial dilated cardiomyopathy (DCM) is clinically variable and has been associated with mutations in more than 50 genes. Rapid improvements in DNA sequencing have led to the identification of diverse rare variants with unknown significance (VUS), which underlines the importance of functional analyses. In this study, by investigating human-induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs), we evaluated the pathogenicity of the p.C335R sodium voltage-gated channel alpha subunit 5 () variant in a large family with familial DCM and conduction disease.
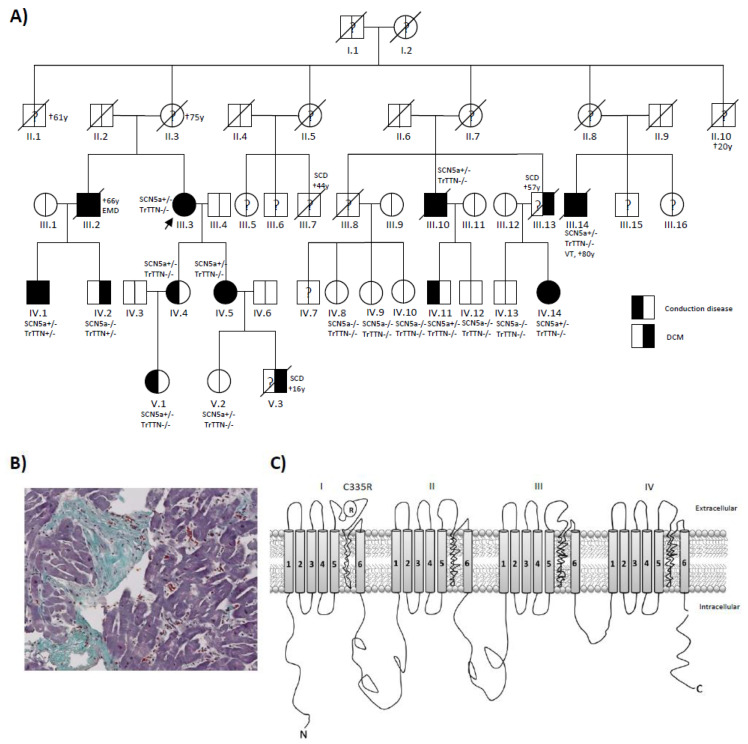
A four-generation family with autosomal dominant familial DCM was investigated. Next-generation sequencing (NGS) was performed in all 16 family members. Clinical deep phenotyping, including endomyocardial biopsy, was performed. Skin biopsies from two patients and one healthy family member were used to generate human-induced pluripotent stem cells (iPSCs), which were then differentiated into cardiomyocytes. Patch-clamp analysis with oocytes and iPSC-CMs were performed.
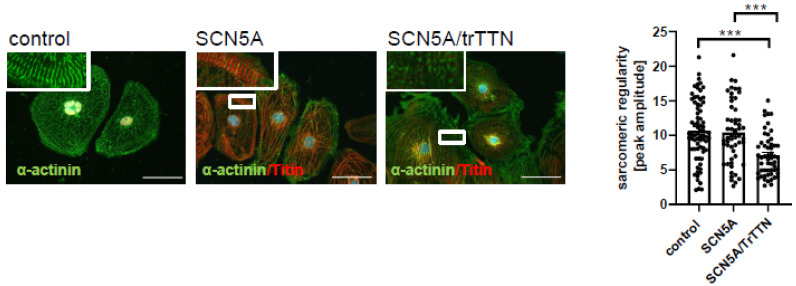
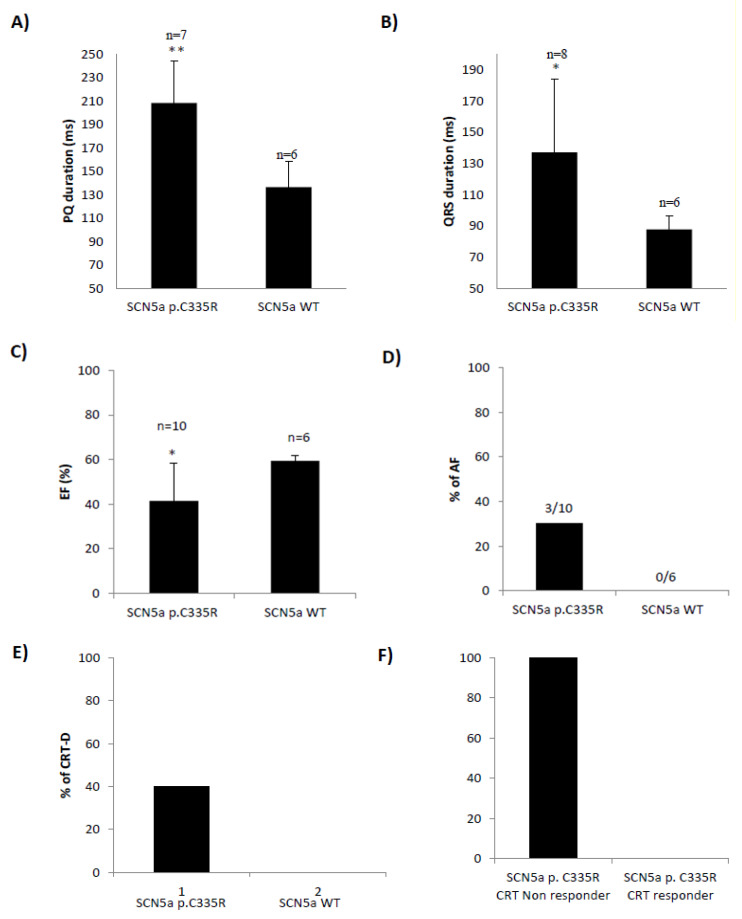
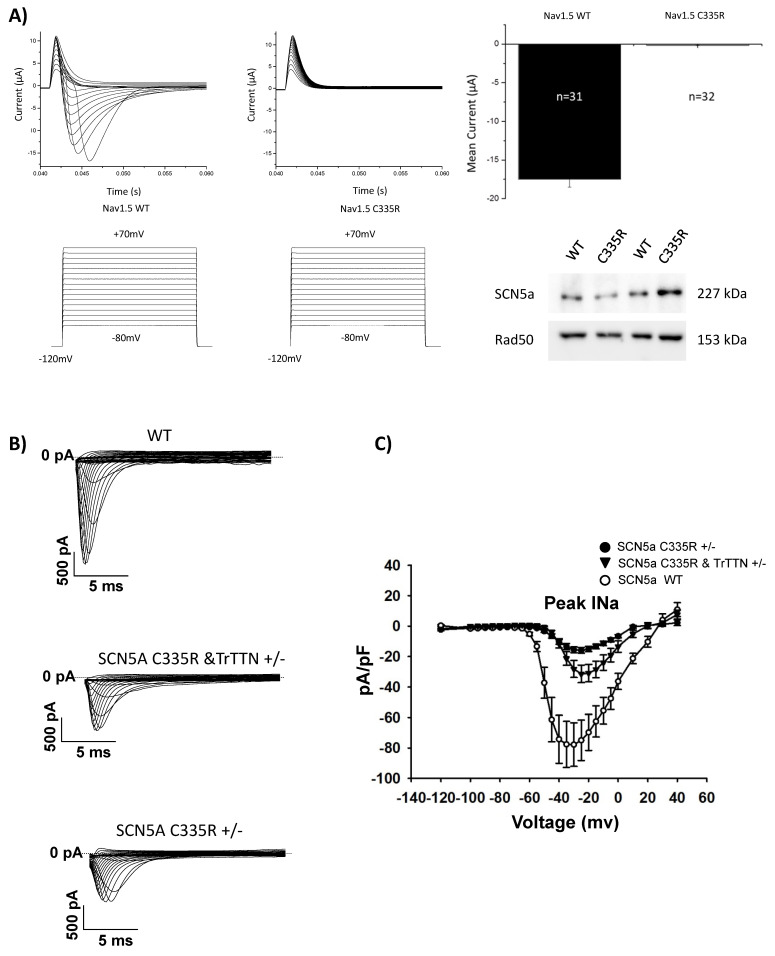
A variant (c.1003T>C; p.C335R) could be detected in all family members with DCM or conduction disease. A novel truncating variant (p.Ser24998LysfsTer28) could also be identified in two family members with DCM. Family members with the variant (p.C335R) showed significantly longer PQ and QRS intervals and lower left ventricular ejection fractions (LV-EF). All four patients who received CRT-D were non-responders. Electrophysiological analysis with oocytes showed a loss of function in p.C335R. Na channel currents were also reduced in iPSC-CMs from DCM patients. Furthermore, iPSC-CM with compound heterozygosity ( p.C335R and ) showed significant dysregulation of sarcomere structures, which may be contributed to the severity of the disease and earlier onset of DCM.
The p.C335R variant is causing a loss of function of peak INa in patients with DCM and cardiac conduction disease. The co-existence of genetic variants in channels and structural genes (e.g., p.C335R and ) increases the severity of the DCM phenotype.
家族性扩张型心肌病(DCM)临床表现多样,与 50 多种基因突变相关。DNA 测序技术的快速发展导致了大量具有未知意义的稀有变异(VUS)的发现,这凸显了功能分析的重要性。在这项研究中,通过研究人类诱导多能干细胞衍生的心肌细胞(iPSC-CMs),我们评估了一个具有家族性 DCM 和传导疾病的大型家族中 p.C335R 钠电压门控通道 alpha 亚单位 5 () 变体的致病性。
研究了一个四代常染色体显性遗传性家族性 DCM 家族。对所有 16 名家族成员进行了下一代测序(NGS)。进行了包括心内膜心肌活检在内的临床深度表型分析。从两名患者和一名健康家族成员的皮肤活检中提取人类诱导多能干细胞(iPSCs),并将其分化为心肌细胞。利用卵母细胞和 iPSC-CMs 进行膜片钳分析。
在所有患有 DCM 或传导疾病的家族成员中均能检测到 p.C335R 变体。在两名患有 DCM 的家族成员中还发现了一种新的截断变体(p.Ser24998LysfsTer28)。携带 p.C335R 变体的家族成员 PQ 和 QRS 间期明显延长,左心室射血分数(LV-EF)降低。所有接受 CRT-D 治疗的四名患者均无反应。卵母细胞电生理分析显示 p.C335R 丧失功能。DCM 患者的 iPSC-CMs 中的钠通道电流也减少了。此外,复合杂合性( p.C335R 和 )的 iPSC-CM 显示肌节结构明显失调,这可能导致疾病的严重程度和 DCM 的发病年龄更早。
p.C335R 变体导致 DCM 和心脏传导疾病患者的峰值 INa 功能丧失。通道和结构基因(例如, p.C335R 和 )中的遗传变异共存增加了 DCM 表型的严重程度。