Nasereddin Abedelmajeed, Ereqat Suheir
Al-Quds Nutrition and Health Research Institute, Faculty of Medicine-Al-Quds University, P.O. Box 20760, Abu-Dis-Esat Jerusalem, Palestine.
Genomics Applications Lab, The Core Research Facility, Faculty of Medicine, The Hebrew University, Jerusalem, Israel.
J Med Case Rep. 2018 Sep 18;12(1):272. doi: 10.1186/s13256-018-1805-x.
Niemann-Pick disease is caused by reduced level of the lysosomal enzyme acid sphingomyelinase. Children can survive between 2 and 12 years based on the disease type. Two main types are well known: type A and B. Niemann-Pick disease type A is characterized by severe central nervous system deterioration and hepatosplenomegaly while type B is a progressive hypersplenism accompanied with gradual deterioration of pulmonary function.
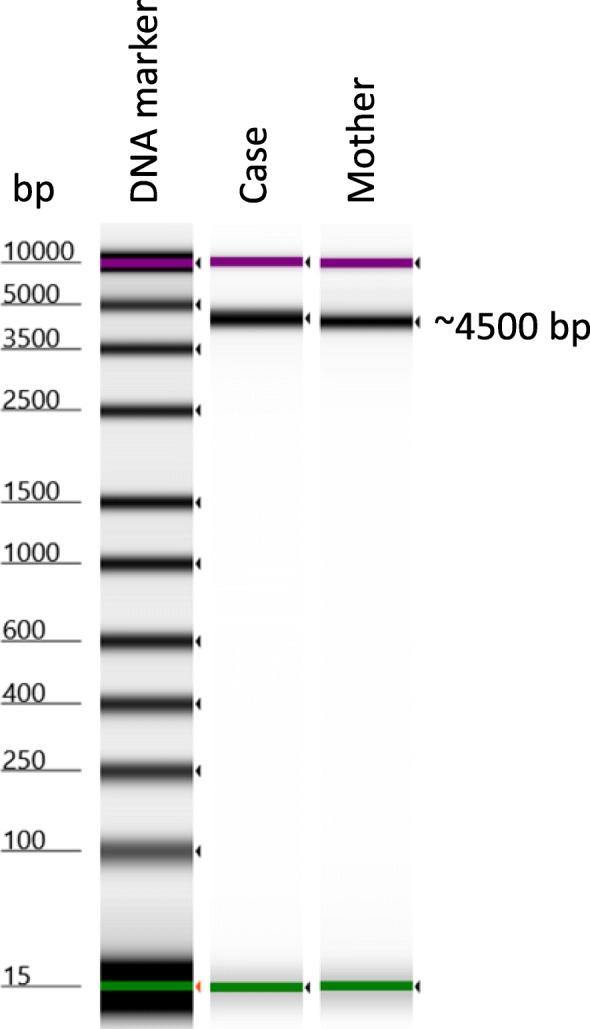
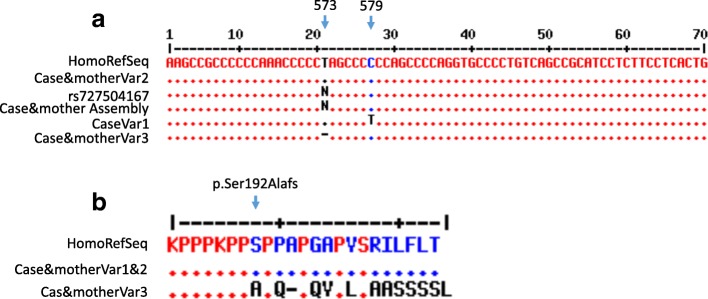
We describe an 11-month-old Palestinian baby boy with hepatosplenomegaly, hypotonia, delayed motor development, laryngomalacia, bilateral cherry-red spots, and failure to thrive. Metabolic screening, blood count, differential tests, immunology screen, infectious disease screen, urine, biochemical tests as well as molecular diagnosis were performed. The molecular diagnosis was done by amplifying the whole sphingomyelin phosphodiesterase 1 (SMPD1) gene, followed by deep sequencing. The obtained sequences were aligned, de novo assembled and compared to human reference gene (GenBank GeneID: NG_011780.1, Ensembl version ENSG00000166311 and protein identified as UniProtKB - P17405). Two known mutations were identified in our patient: the pathogenic frameshift mutation NM_000543.4(SMPD1):c.573delT (p.Ser192Alafs) and the benign polymorphism NM_000543.4(SMPD1):c.107T>C (p.Val36Ala). The enzyme study showed a very low level of enzymatic activity of acidic sphingomyelinase (0.1 nmol/ml per hour). Correlations between clinical findings, laboratory data, and sequence analysis are presented.
In conclusion, this is the first report about a heterozygote frameshift p.Ser192AlafsX65 in a Palestinian patient with Niemann-Pick disease type A, emphasizing the importance of deep sequencing in genetic diagnosis of this rare inherited disease.
尼曼-匹克病是由溶酶体酶酸性鞘磷脂酶水平降低引起的。根据疾病类型,患儿的存活时间在2至12岁之间。已知有两种主要类型:A型和B型。尼曼-匹克病A型的特征是严重的中枢神经系统恶化和肝脾肿大,而B型是进行性脾功能亢进并伴有肺功能逐渐恶化。
我们描述了一名11个月大的巴勒斯坦男婴,有肝脾肿大、肌张力减退、运动发育迟缓、喉软化、双侧樱桃红斑和发育不良。进行了代谢筛查、血常规、分类检查、免疫学筛查、传染病筛查、尿液、生化检查以及分子诊断。分子诊断通过扩增整个鞘磷脂磷酸二酯酶1(SMPD1)基因,然后进行深度测序来完成。将获得的序列进行比对、从头组装并与人类参考基因(GenBank基因ID:NG_011780.1,Ensembl版本ENSG00000166311,蛋白质鉴定为UniProtKB - P17405)进行比较。在我们的患者中鉴定出两个已知突变:致病的移码突变NM_000543.4(SMPD1):c.573delT(p.Ser192Alafs)和良性多态性NM_(p.Val36Ala)。酶学研究显示酸性鞘磷脂酶的酶活性水平非常低(每小时0.1 nmol/ml)。呈现了临床发现、实验室数据和序列分析之间的相关性。
总之,这是关于一名患有尼曼-匹克病A型的巴勒斯坦患者中杂合子移码p.Ser192AlafsX65的首次报告,强调了深度测序在这种罕见遗传病基因诊断中的重要性。