Institute of Biomedical and Clinical Science, University of Exeter Medical School, Exeter, UK.
Paediatric Department, Hospital Putrajaya, Putrajaya, Malaysia.
Clin Endocrinol (Oxf). 2018 Nov;89(5):621-627. doi: 10.1111/cen.13841. Epub 2018 Sep 20.
Hyperinsulinaemic hypoglycaemia (HH) can occur in isolation or more rarely feature as part of a syndrome. Screening for mutations in the "syndromic" HH genes is guided by phenotype with genetic testing used to confirm the clinical diagnosis. As HH can be the presenting feature of a syndrome, it is possible that mutations will be missed as these genes are not routinely screened in all newly diagnosed individuals. We investigated the frequency of pathogenic variants in syndromic genes in infants with HH who had not been clinically diagnosed with a syndromic disorder at referral for genetic testing.
We used genome sequencing data to assess the prevalence of mutations in syndromic HH genes in an international cohort of patients with HH of unknown genetic cause.
We undertook genome sequencing in 82 infants with HH without a clinical diagnosis of a known syndrome at referral for genetic testing.
Within this cohort, we searched for the genetic aetiologies causing 20 different syndromes where HH had been reported as a feature.
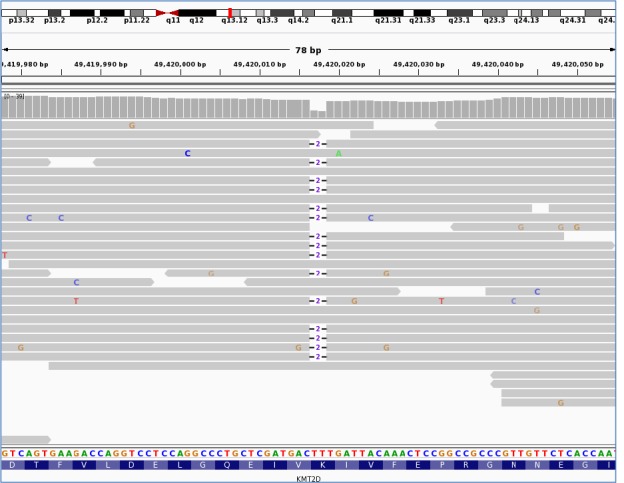
We identified a pathogenic KMT2D variant in a patient with HH diagnosed at birth, confirming a genetic diagnosis of Kabuki syndrome. Clinical data received following the identification of the mutation highlighted additional features consistent with the genetic diagnosis. Pathogenic variants were not identified in the remainder of the cohort.
Pathogenic variants in the syndromic HH genes are rare; thus, routine testing of these genes by molecular genetics laboratories is unlikely to be justified in patients without syndromic phenotypes.
高胰岛素血症性低血糖(HH)可单独发生,也可作为综合征的一部分更罕见地出现。“综合征性”HH 基因的突变筛查由表型指导,基因检测用于确认临床诊断。由于 HH 可能是综合征的首发特征,因此,由于这些基因在所有新诊断的个体中并非常规筛查,因此可能会错过突变。我们研究了未在转诊进行基因检测时临床诊断为综合征的 HH 婴儿中综合征基因中致病性变异的频率。
我们使用基因组测序数据评估了具有未知遗传病因的 HH 患者国际队列中综合征性 HH 基因中突变的流行率。
我们对 82 名无已知综合征临床诊断的 HH 婴儿进行了基因组测序,这些婴儿在转诊进行基因检测时。
在该队列中,我们在报告 HH 为特征的 20 种不同综合征中寻找导致其发生的遗传病因。
我们在一名出生时即被诊断为 HH 的患者中发现了致病性 KMT2D 变异,从而确诊了卡布基综合征的遗传诊断。在识别出突变后收到的临床数据突出了与遗传诊断一致的其他特征。在其余患者中未发现致病性变异。
综合征性 HH 基因中的致病性变异很少见;因此,在没有综合征表型的患者中,分子遗传学实验室对这些基因进行常规检测不太可能合理。