MitoCare Center, Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University, Philadelphia, PA, USA.
Cell Death Dis. 2018 Oct 9;9(10):1028. doi: 10.1038/s41419-018-1070-3.
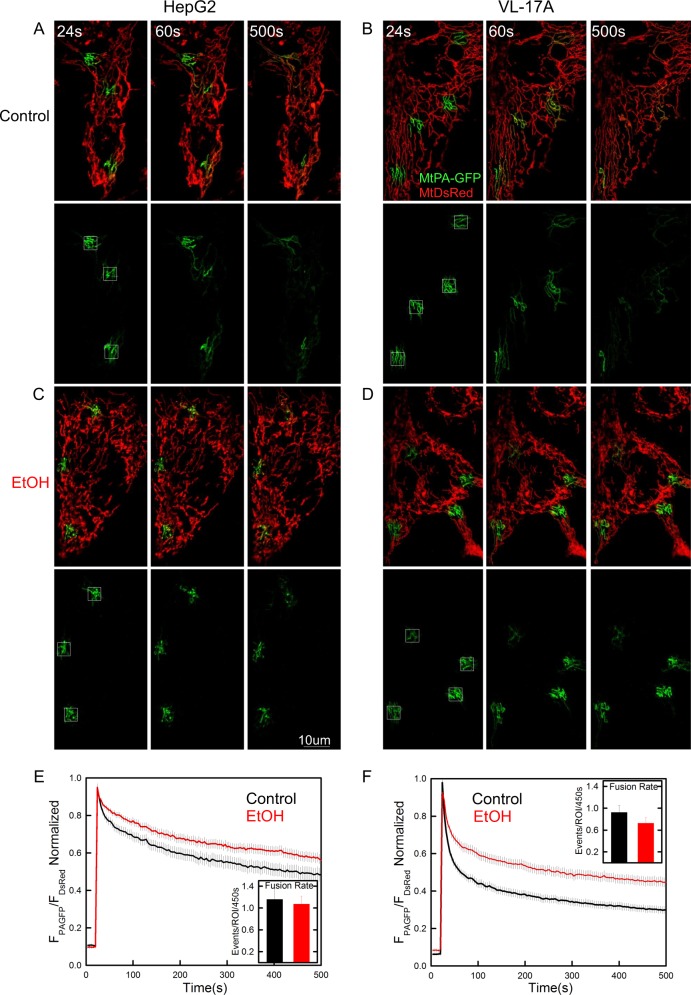
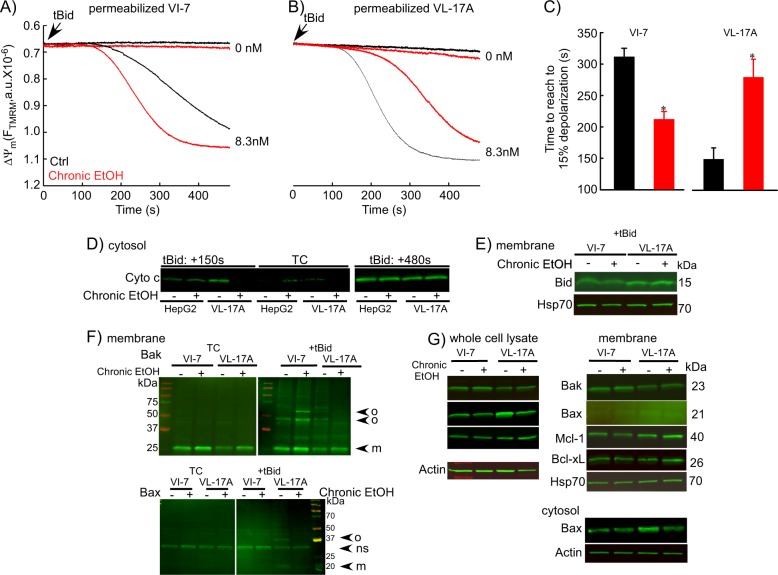
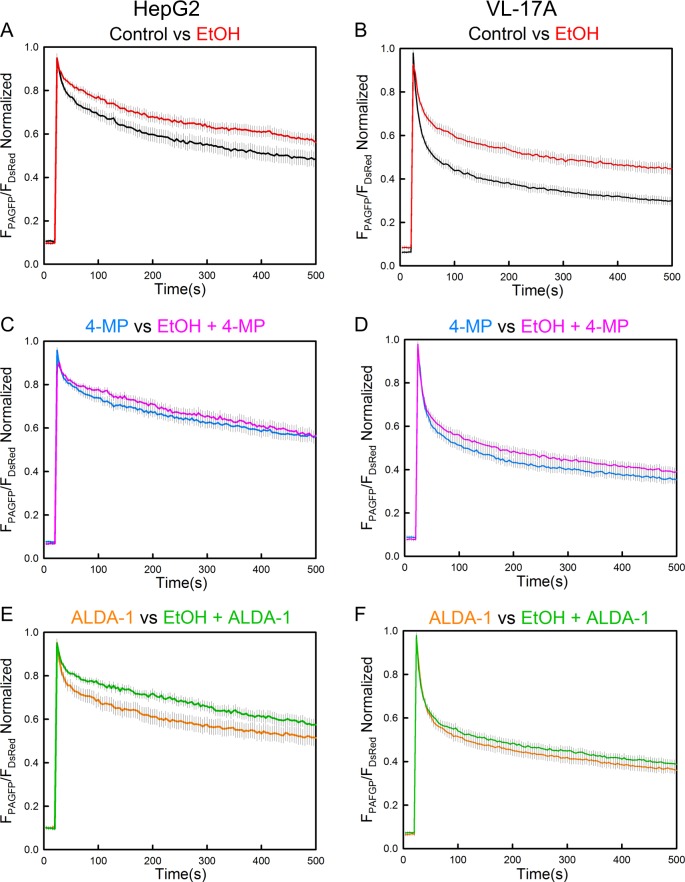
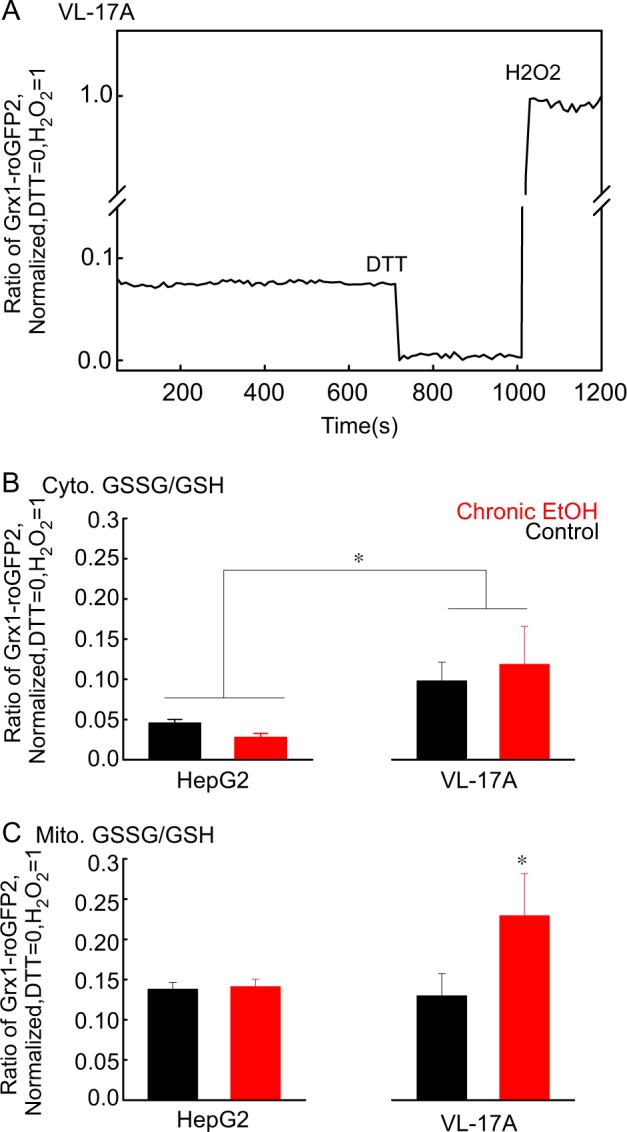
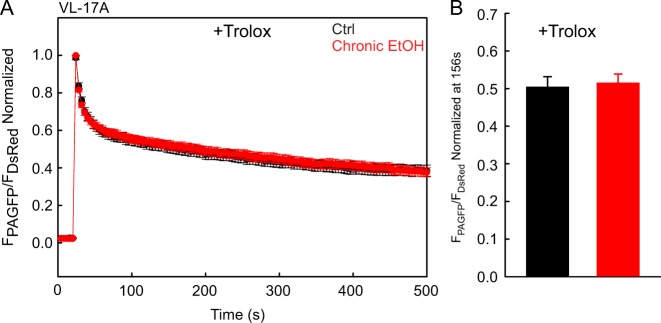
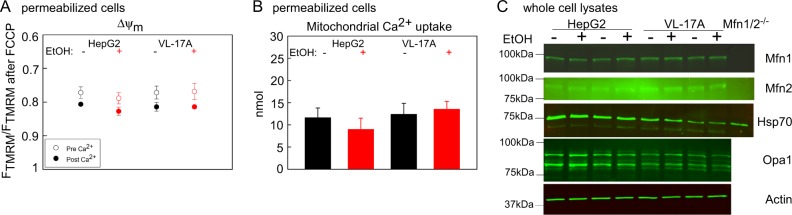
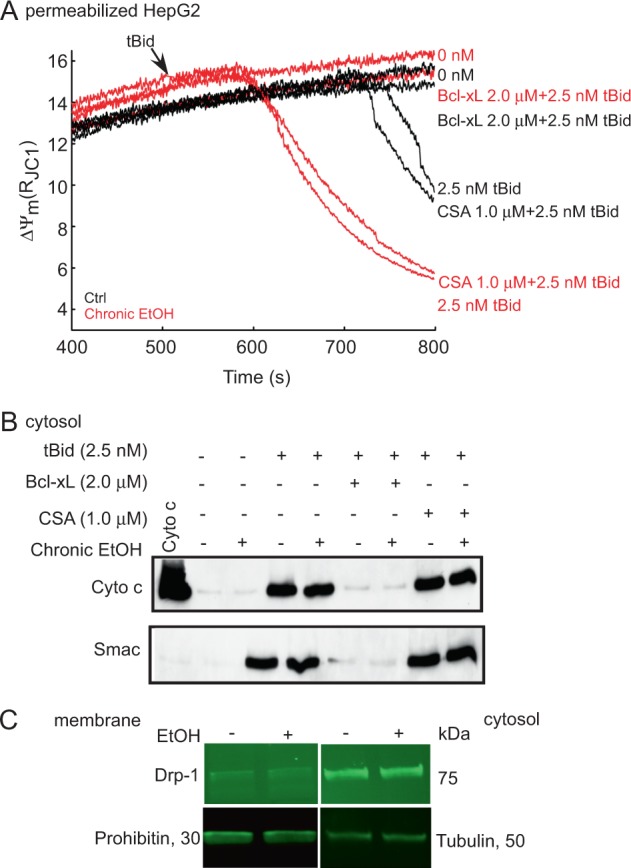
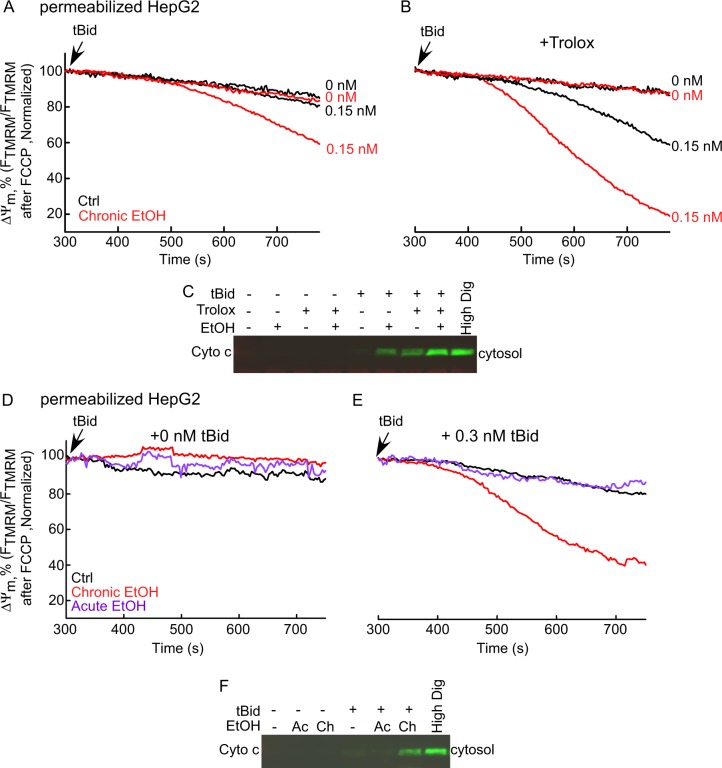
Environmental stressors like ethanol (EtOH) commonly target mitochondria to influence the cell's fate. Recent literature supports that chronic EtOH exposure suppresses mitochondrial dynamics, central to quality control, and sensitizes mitochondrial permeability transition pore opening to promote cell death. EtOH-induced tissue injury is primarily attributed to its toxic metabolic products but alcoholism also impairs tissues that poorly metabolize EtOH. We embarked on studies to determine the respective roles of EtOH and its metabolites in mitochondrial fusion and tBid-induced mitochondrial apoptosis. We used HepG2 cells that do not metabolize EtOH and its engineered clone that expresses EtOH-metabolizing Cytochrome P450 E2 and alcohol dehydrogenase (VL-17A cells). We found that fusion impairment by prolonged EtOH exposure was prominent in VL-17A cells, probably owing to reactive oxygen species increase in the mitochondrial matrix. There was no change in fusion protein abundance, mitochondrial membrane potential or Ca uptake. By contrast, prolonged EtOH exposure promoted tBid-induced outer mitochondrial membrane permeabilization and cell death only in HepG2 cells, owing to enhanced Bak oligomerization. Thus, mitochondrial fusion inhibition by EtOH is dependent on its metabolites, whereas sensitization to tBid-induced death is mediated by EtOH itself. This difference is of pathophysiological relevance because of the tissue-specific differences in EtOH metabolism.
环境应激物,如乙醇(EtOH),通常以线粒体为靶点,影响细胞命运。最近的文献支持慢性 EtOH 暴露抑制线粒体动力学,这是质量控制的核心,并使线粒体通透性转换孔开放敏感,以促进细胞死亡。EtOH 引起的组织损伤主要归因于其有毒代谢产物,但酗酒也会损害代谢 EtOH 能力差的组织。我们着手研究确定 EtOH 及其代谢物在线粒体融合和 tBid 诱导的线粒体凋亡中的各自作用。我们使用不代谢 EtOH 的 HepG2 细胞和表达 EtOH 代谢细胞色素 P450 E2 和醇脱氢酶的工程克隆(VL-17A 细胞)。我们发现,延长 EtOH 暴露导致融合受损在 VL-17A 细胞中更为明显,可能是由于线粒体基质中活性氧增加所致。融合蛋白丰度、线粒体膜电位或 Ca 摄取没有变化。相比之下,延长 EtOH 暴露仅在 HepG2 细胞中促进 tBid 诱导的外线粒体膜通透性和细胞死亡,这是由于 Bak 寡聚化增强所致。因此,EtOH 对线粒体融合的抑制依赖于其代谢物,而对 tBid 诱导的死亡的敏感性则由 EtOH 本身介导。这种差异具有病理生理学相关性,因为 EtOH 代谢在组织特异性方面存在差异。