Ley-Martos Myriam, Guerrero Juan M, Lucas-Javato Marta, Remón-García Cristina, García-Lozano J Raúl, Colón Cristóbal, Crujeiras Pablo, Rodrigues Daniel, Paúl-Sánchez Pedro, Macher Hada C
Pediatric Neurology and Rare Diseases Unit, Department of Pediatry, Hospital Universitario Puerta del Mar, Cádiz.
Molecular Diagnosis and Rare Diseases Laboratory, Department of Clinical Chemistry, Hospital Universitario Virgen del Rocío and Instituto de Biomedicina de Sevilla (IBiS), Sevilla.
Medicine (Baltimore). 2018 Oct;97(42):e12872. doi: 10.1097/MD.0000000000012872.
Mucopolysaccharidosis type VI (MPS VI) or Maroteaux-Lamy syndrome is produced by the deficiency of the enzyme arylsulfatase B, responsible for the hydrolysis of N-acetyl-D-galactosamine, chondroitin sulfate, and dermatan sulfate.
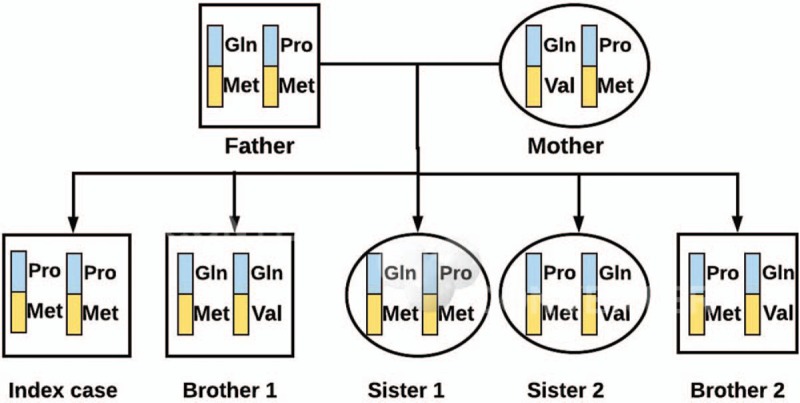
A 3-year-old male with Moroccan origins is the index case. He had healthy consanguineous parents and 4 healthy brothers and sisters. The patient showed a wide spectrum of symptoms including skeletal dysplasia and short stature with elevated glycosaminoglycans (GAGs) in urine.
DIAGNOSES, INTERVENTIONS, AND OUTCOMES: GAGs were quantified by spectrometry method with 1,9-dimethylen blue in 24-hour urine samples. The qualitative analysis of urine GAGs was obtained by thin-layer chromatography to determine the predominant presence of dermatan sulfate. The activities of both arylsulfatase B and beta-galactosidase as well as genetic studies were performed in dried blood spots. The genetic study was performed with deoxyribonucleic acid by massive sequencing a of lisosomal storage diseases. Results showed a new mutation c.263A > C with the severe phenotype in homozygous in the patient. The familiar study of ARSB and GLB1 genes presented some asymptomatic SNPs but with a discrete decrease in the activity of arylsulfatase B and beta-galactosidase. After an early detection by pediatricians, and both enzymatic and genetic confirmation, the patient had a good response to substitutive enzymatic treatment with galsulfase.
Mucoplysaccharidosis type VI is an autosomal recessive rare disease characterized by a lysosomal storage disorder. Although a number of mutations have been already associated to the disease, we have found a new mutation located in the arylsulfatase B enzyme gene. We have described that this mutation is the ultimate cause of a severe presentation of the disease.
VI型黏多糖贮积症(MPS VI)或马罗-拉米综合征是由芳基硫酸酯酶B缺乏引起的,该酶负责N-乙酰-D-半乳糖胺、硫酸软骨素和硫酸皮肤素的水解。
一名3岁摩洛哥裔男性为索引病例。他的父母近亲结婚但身体健康,还有4个健康的兄弟姐妹。该患者表现出广泛的症状,包括骨骼发育异常和身材矮小,尿中糖胺聚糖(GAGs)升高。
诊断、干预及结果:采用1,9-二亚甲基蓝分光光度法对24小时尿液样本中的GAGs进行定量。通过薄层色谱法对尿GAGs进行定性分析,以确定硫酸皮肤素的主要存在情况。在干血斑中进行芳基硫酸酯酶B和β-半乳糖苷酶的活性检测以及基因研究。通过对溶酶体贮积病进行大规模测序,用脱氧核糖核酸进行基因研究。结果显示患者存在一个新的纯合突变c.263A>C,具有严重表型。对ARSB和GLB1基因的家系研究发现了一些无症状的单核苷酸多态性,但芳基硫酸酯酶B和β-半乳糖苷酶的活性有一定程度降低。在儿科医生早期诊断以及酶学和基因确认后,患者对用加尔硫酸酶进行的替代酶治疗反应良好。
VI型黏多糖贮积症是一种常染色体隐性罕见疾病,其特征为溶酶体贮积障碍。尽管已经发现了许多与该疾病相关的突变,但我们发现了一个位于芳基硫酸酯酶B酶基因中的新突变。我们已经证明该突变是该疾病严重表现的最终原因。