Ye Fanghua, Chai Wenwen, Yang Minghua, Xie Min, Yang Liangchun
Department of Pediatrics, Xiangya Hospital, Central South University, Changsha, Hunan 410008, P.R. China.
Department of Nuclear Medicine, Hunan Cancer Hospital and The Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha, Hunan 410008, P.R. China.
Mol Clin Oncol. 2018 Nov;9(5):493-498. doi: 10.3892/mco.2018.1721. Epub 2018 Sep 17.
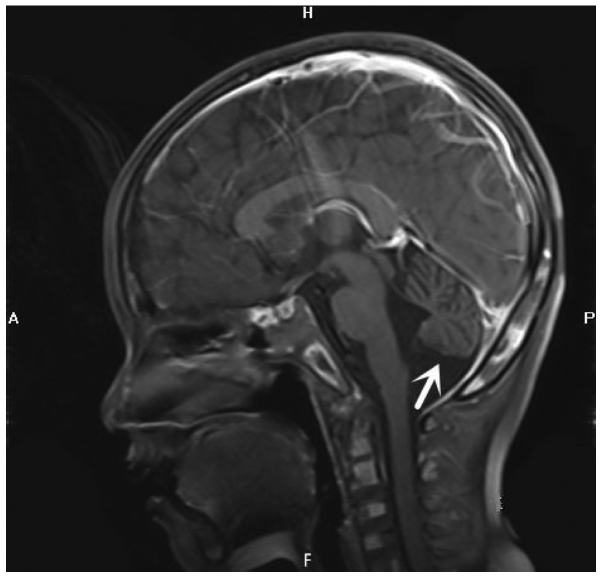
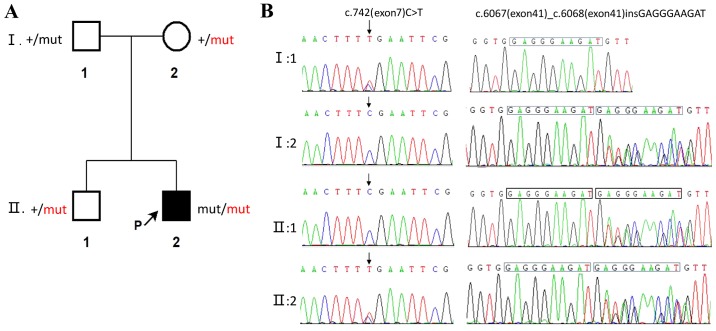
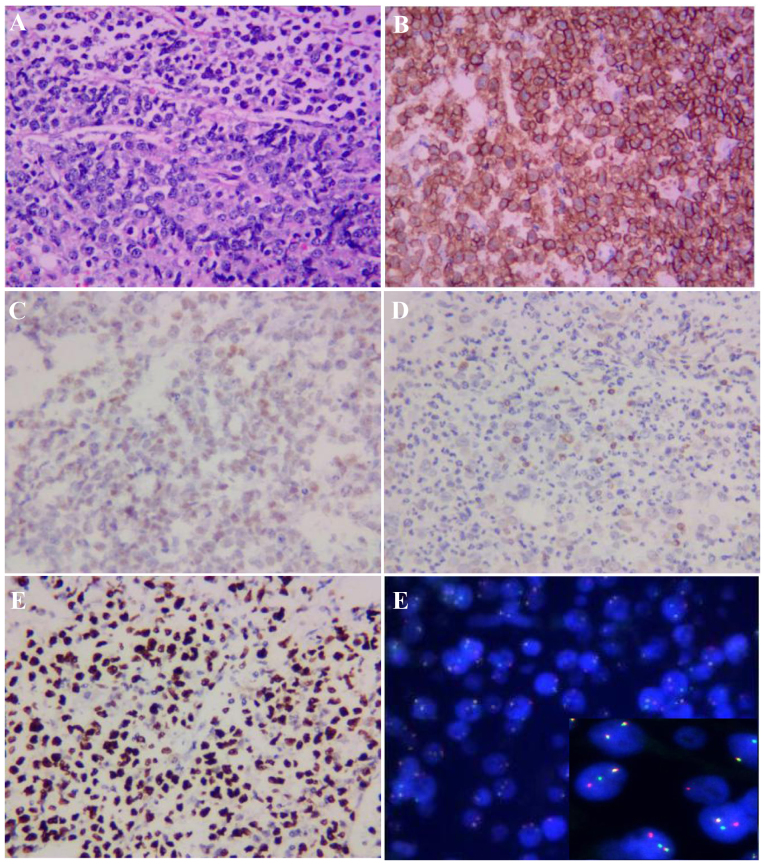
Ataxia-telangiectasia (A-T) is an infrequent autosomal recessive disorder that involves multiple systems and is characterized by progressive cerebellar ataxia, oculocutaneous telangiectasias, radiosensitivity, immune deficiency with recurrent respiratory infections, and a tendency to develop lymphoid malignancies. A-T is caused by mutations in the ATM gene, with >1,000 mutations reported to date and gradually increasing in number. Patients with A-T have an increased incidence of cancers. The aim of the present study was to retrospectively review the case of a patient who presented at the age of 5 years with cerebellar ataxia without telangiectasia, and was diagnosed with Burkitt leukemia by bone marrow biopsy and molecular testing at the age of 7 years at the Xiangya Hospital of Central South University (Changsha, China). The patient received chemotherapy with the pediatric CCCG-BNHL-2015 regimen (R4 group) and achieved a complete remission after 2 courses. However, recurrent respiratory infections and thrombosis occurred during chemotherapy. The diagnosis of A-T was confirmed by uncovering two variants of the ATM gene, including c.742C>T (p.R248X; rs730881336) in exon 7 and c.6067-c.6068 ins GAGGGAAGAT in exon 41 by whole-exome sequencing. Unfortunately, the patient's parents refused follow-up treatment and he succumbed to recurrent severe infections 4 months after the diagnosis of Burkitt leukemia. The diagnosis of A-T may be challenging, as its phenotype can be incomplete early in the course of the disease. Detailed medical history, characteristic clinical manifestations and increasingly developed exome sequencing techniques may be helpful in diagnosing this rare disease. Management should be based on multidisciplinary guidance and other treatment options must be investigated in the future.
共济失调毛细血管扩张症(A-T)是一种罕见的常染色体隐性疾病,累及多个系统,其特征为进行性小脑共济失调、眼皮肤毛细血管扩张、放射敏感性、免疫缺陷伴反复呼吸道感染以及发生淋巴系统恶性肿瘤的倾向。A-T由ATM基因突变引起,迄今已报道1000多种突变,且数量在逐渐增加。A-T患者患癌症的几率增加。本研究的目的是回顾性分析一名5岁时出现无毛细血管扩张的小脑共济失调、7岁时在中南大学湘雅医院(中国长沙)经骨髓活检和分子检测诊断为伯基特白血病的患者病例。该患者接受了小儿CCCG-BNHL-2015方案(R4组)化疗,2个疗程后实现完全缓解。然而,化疗期间出现了反复呼吸道感染和血栓形成。通过全外显子组测序发现ATM基因的两个变异,包括外显子7中的c.742C>T(p.R248X;rs730881336)和外显子41中的c.6067-c.6068插入GAGGGAAGAT,从而确诊为A-T。不幸的是,患者父母拒绝后续治疗,他在伯基特白血病诊断4个月后死于反复严重感染。A-T的诊断可能具有挑战性,因为其表型在疾病早期可能不完整。详细的病史、特征性临床表现以及日益发展的外显子组测序技术可能有助于诊断这种罕见疾病。治疗应基于多学科指导,未来必须探索其他治疗选择。