Iwasaki Yasushi, Hashimoto Rina, Saito Yufuko, Aiba Ikuko, Inukai Akira, Akagi Akio, Mimuro Maya, Miyahara Hiroaki, Kitamoto Tetsuyuki, Yoshida Mari
a Department of Neuropathology, Institute for Medical Science of Aging , Aichi Medical University , Nagakute , Japan.
b Department of Neurology , National Hospital Organization Higashinagoya National Hospital , Nagoya , Japan.
Prion. 2019 Jan;13(1):13-20. doi: 10.1080/19336896.2018.1545525. Epub 2018 Nov 14.
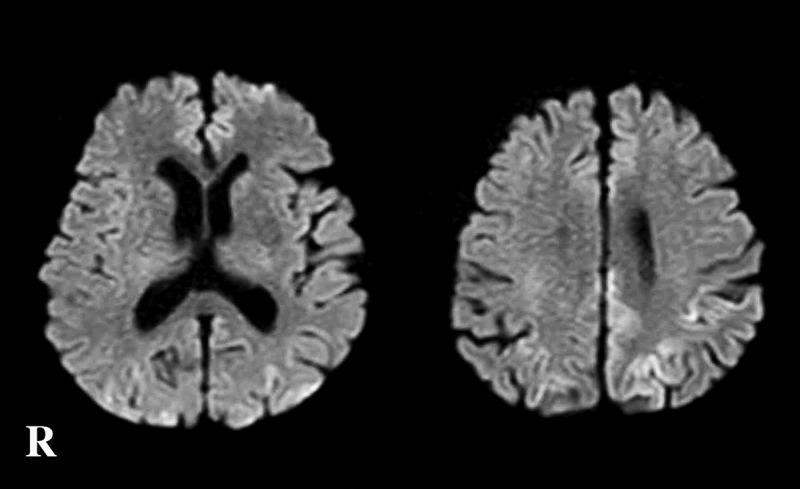
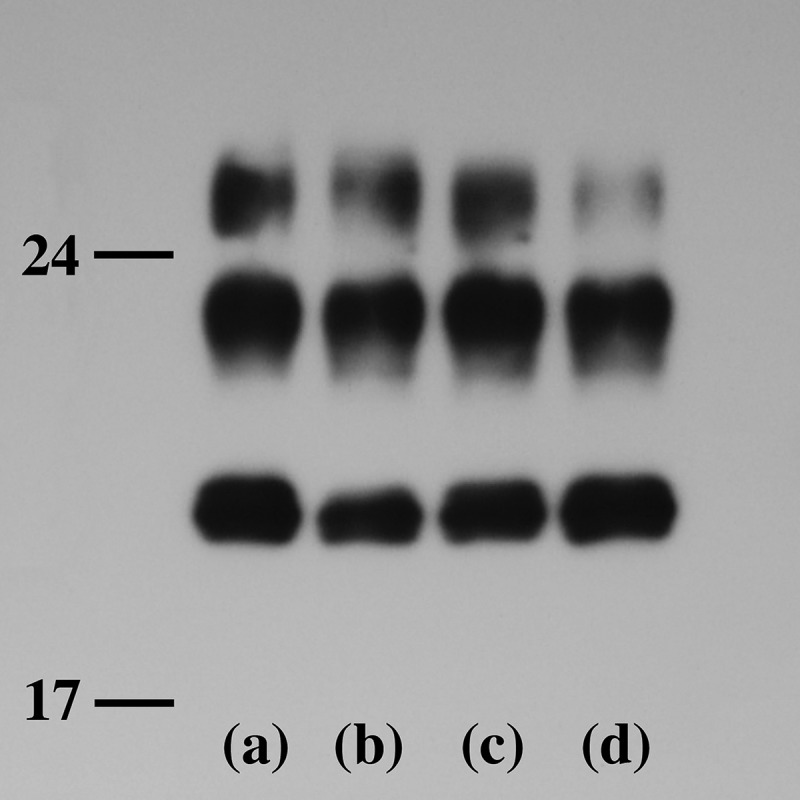
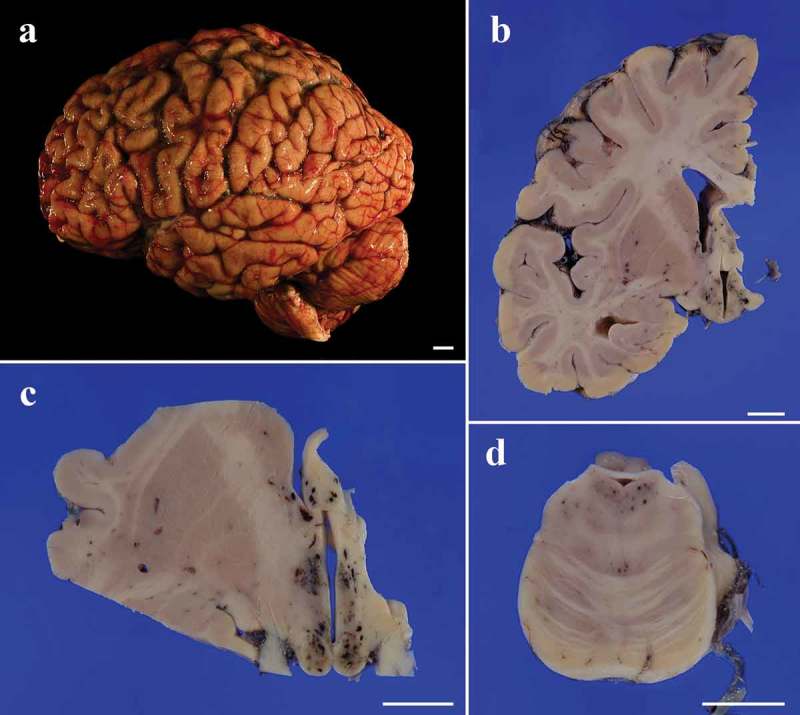
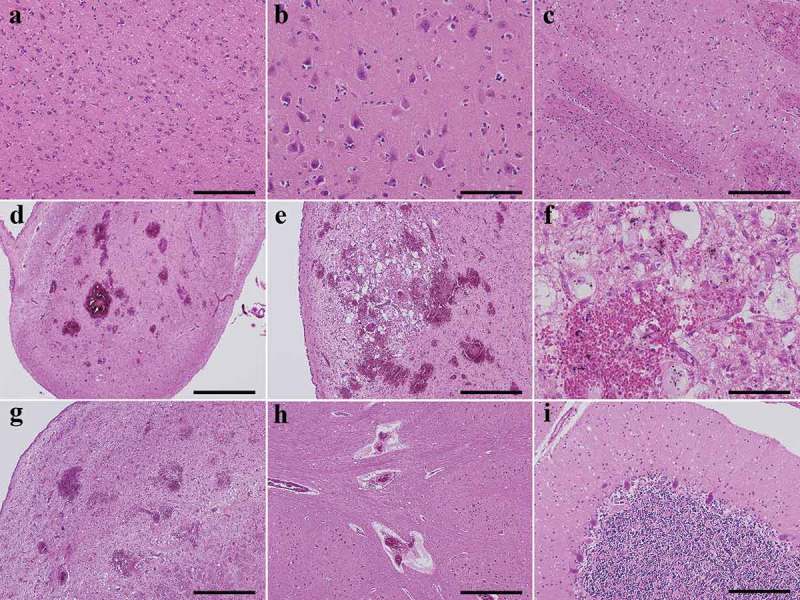
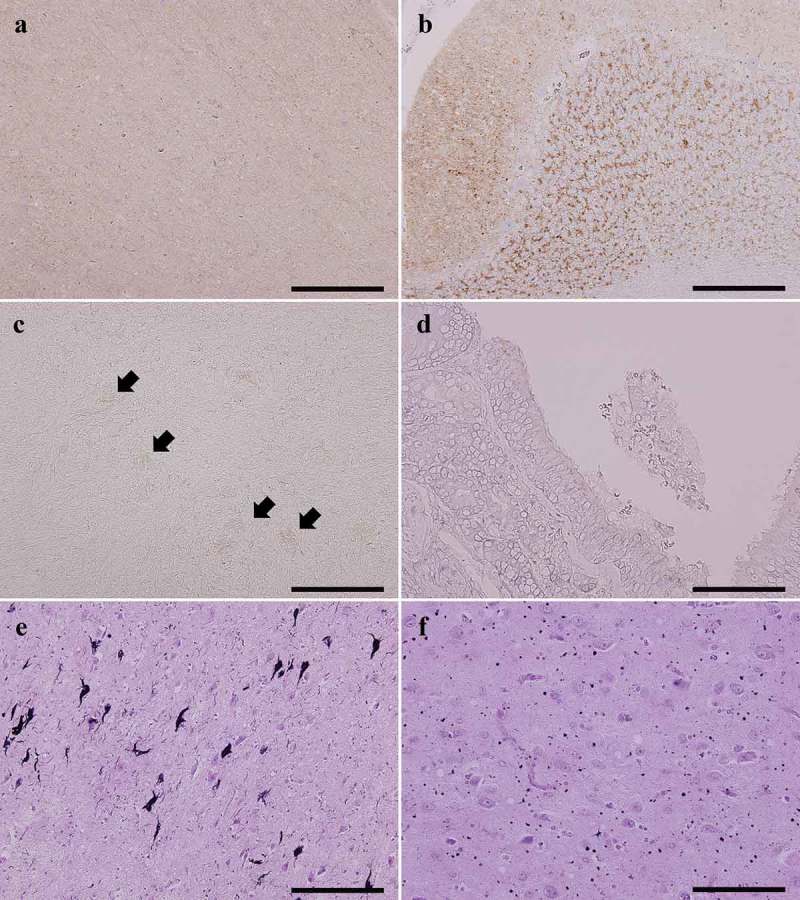
An 83-year-old Japanese man presented with gait disturbance followed by rapidly-progressive cognitive impairment. Magnetic resonance diffusion-weighted images showed extensive hyperintense regions in the cerebral cortex. Four weeks after symptom onset, myoclonus appeared, and the patient developed difficulty swallowing; intravenous peripheral continuous infusions without vitamin supplementation were administered during the last two months of the patient's life. The patient reached the akinetic mutism state and died 12 weeks after symptom onset due to sepsis. The brain weighed 940 g and showed general cerebral atrophy. Extensive spongiform change were observed in the cerebral cortex, striatum, thalamus, and cerebellar cortex, but gliosis was generally mild. Numerous newly-developed hemorrhage foci were observed in the mammillary body, the areas adjacent to the third and fourth ventricles, and the periaqueduct of the midbrain; however, proliferation of capillaries and endothelium and collections of macrophages were relatively inconspicuous. These findings suggested comorbidity with the acute stage of Wernicke encephalopathy (WE). Immunostaining showed extensive diffuse synaptic-type prion protein deposition in the gray matter. According to the neuropathological, genetic, and molecular findings, the present case was finally diagnosed as MM1-type sporadic Creutzfeldt-Jakob disease (CJD) with WE. We should remain alert to the diagnosis of WE when CJD is suspected, and it is necessary to consider the complications of both diseases. This report emphasizes the importance of pathological investigations for the diagnosis of CJD, WE, and the coexistence of both.
一名83岁的日本男性出现步态障碍,随后迅速出现进行性认知障碍。磁共振扩散加权成像显示大脑皮层有广泛的高信号区域。症状出现四周后,出现肌阵挛,患者出现吞咽困难;在患者生命的最后两个月,在未补充维生素的情况下进行了静脉外周持续输注。患者进入运动不能性缄默状态,症状出现12周后因败血症死亡。大脑重量为940克,显示出一般性脑萎缩。在大脑皮层、纹状体、丘脑和小脑皮层观察到广泛的海绵状改变,但胶质细胞增生一般较轻。在乳头体、第三和第四脑室相邻区域以及中脑导水管周围观察到大量新出现的出血灶;然而,毛细血管和内皮细胞的增殖以及巨噬细胞的聚集相对不明显。这些发现提示与韦尼克脑病(WE)急性期合并存在。免疫染色显示灰质中有广泛的弥漫性突触型朊蛋白沉积。根据神经病理学、遗传学和分子学发现,本病例最终诊断为MM1型散发性克雅氏病(CJD)合并WE。当怀疑患有CJD时,我们应警惕WE的诊断,有必要考虑两种疾病的并发症。本报告强调了病理检查对CJD、WE以及两者并存诊断的重要性。