Pediatric Highly Intensive Care Unit, Department of Pathophysiology and Transplantation, Università degli Studi di Milano, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Clinica Pediatrica, Fondazione MBBM, Università Milano-Bicocca, Monza, Italy.
Ital J Pediatr. 2018 Nov 16;44(Suppl 2):128. doi: 10.1186/s13052-018-0566-x.
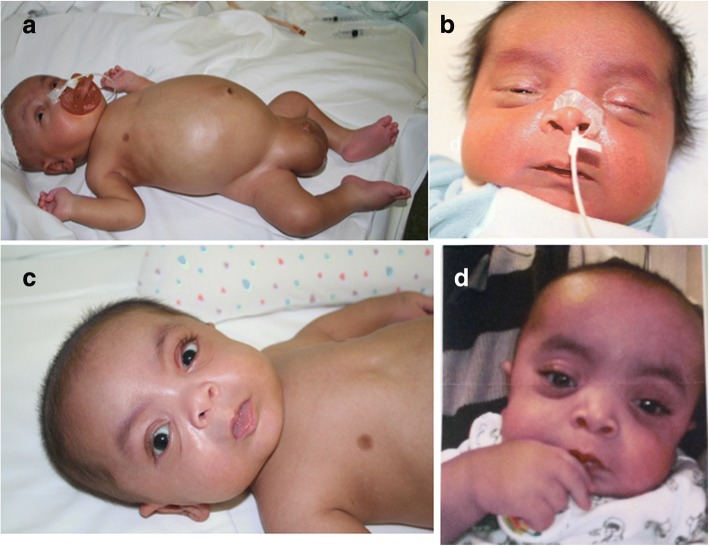
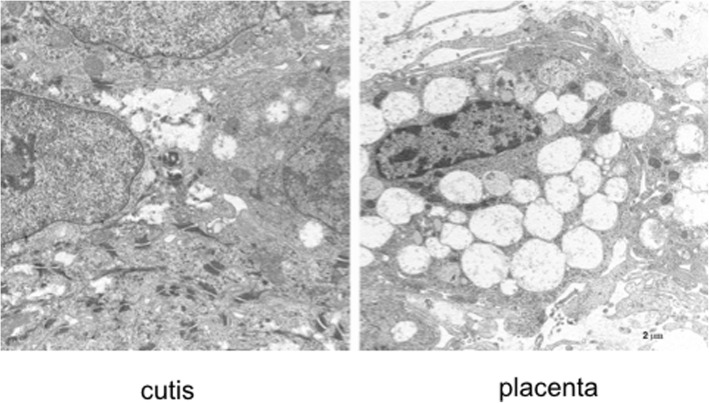
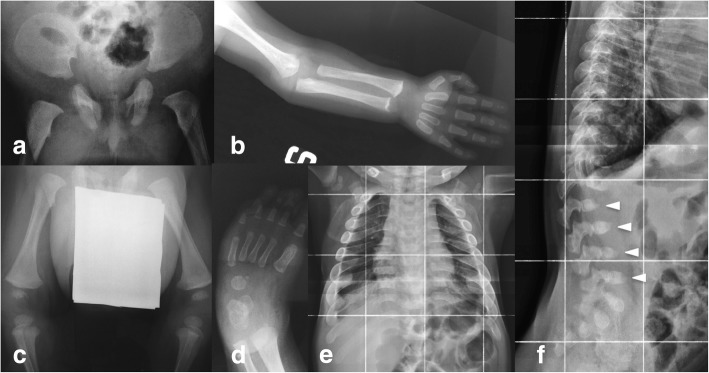
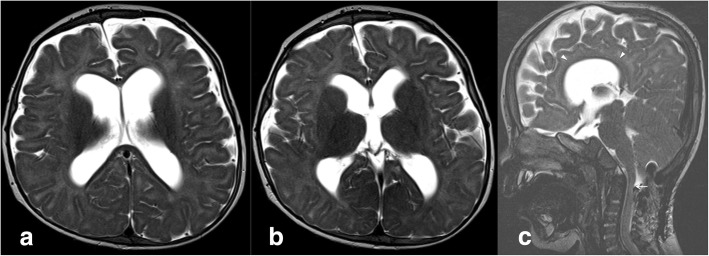
A new patient with severe mucopolysaccharidosis (MPS) type VII is reported. Non-immune hydrops fetalis (NIHF) was diagnosed during pregnancy. At birth, he showed generalized hydrops and dysmorphic features typical of MPS. Many diagnoses were excluded before reaching the diagnosis of MPS VII at 8 months of life. During the first year of life he had frequent respiratory infections associated with restrictive and obstructive bronchopneumopathy and underwent three surgical interventions: decompression of the spinal cord at the craniocervical junction, bilateral inguinal hernia, and bilateral clubfoot. At 14 months of life he underwent successful haematopoietic cell transplantation (HCT). During the following 10 months, his bronchopneumopathy progressively worsened, needing chronic pharmacological treatment and O administration. The patient died of respiratory insufficiency during a respiratory syncytial virus infection at 25 months of age. Molecular analysis showed the homozygous variant c.1617C > T, leading to the synonymous mutation p.Ser539=. This caused aberrant splicing with partial skipping of exon 10 (r.1616_1653del38) and complete skipping of exon 9 (r.1392_1476del85; r.1616_1653del38). No transcript of normal size was evident. The parents were both confirmed to be carriers. In a subsequent pregnancy, a prenatal diagnosis showed an affected fetus. Ultrasound examination before abortion showed NIHF. The skin and placenta examination by electron microscopy showed foamy intracytoplasmic vacuoles with a weakly electron-dense substrate. MPS VII is a very rare disease but it is possible that some cases go undiagnosed for several reasons, including that MPS VII, and other lysosomal storage diseases, are not included in the work-up for NIHF in many institutions, and the presence of anasarca at birth may be confounding for the recognition of the typical facial characteristics of the disease. This is the eighth patient affected by MPS VII who has undergone HCT. It is not possible to draw conclusions about the efficacy of HCT in MPS VII. Treatment with enzyme replacement is now available and will probably be beneficial for the patients who have a milder form with no or little cognitive involvement. Increased awareness among clinicians is needed for prompt diagnosis and to offer the correct treatment as early as possible.
报道了一例新的严重黏多糖贮积症 VII 型(MPS VII)患者。该患者在妊娠期间被诊断为非免疫性胎儿水肿(NIHF)。出生时,他表现出典型的 MPS 全身水肿和畸形特征。在 8 个月大时被确诊为 MPS VII 之前,排除了许多其他诊断。在生命的第一年,他经常发生呼吸道感染,伴有限制性和阻塞性支气管肺炎,并接受了三次手术干预:颅颈交界区脊髓减压、双侧腹股沟疝和双侧马蹄内翻足。14 个月大时,他成功接受了造血细胞移植(HCT)。在接下来的 10 个月里,他的支气管肺炎逐渐恶化,需要长期药物治疗和氧气治疗。25 个月大时,他因呼吸道合胞病毒感染导致呼吸功能衰竭而死亡。分子分析显示纯合变异 c.1617C>T,导致同义突变 p.Ser539=。这导致外显子 10 部分缺失(r.1616_1653del38)和外显子 9 完全缺失(r.1392_1476del85;r.1616_1653del38)的异常剪接,没有明显大小的正常转录本。父母均被确认为携带者。在随后的一次妊娠中,产前诊断显示胎儿受累。流产前的超声检查显示 NIHF。皮肤和胎盘的电子显微镜检查显示有空泡状胞质内空泡,电子密度较弱的基质。MPS VII 是一种非常罕见的疾病,但由于多种原因,一些病例可能未被诊断出来,包括 MPS VII 和其他溶酶体贮积病不在许多机构 NIHF 检查范围内,出生时出现全身水肿可能会干扰对疾病典型面部特征的识别。这是第八例接受 HCT 治疗的 MPS VII 患者。目前还无法得出关于 HCT 在 MPS VII 中的疗效的结论。现在有酶替代治疗,对于没有或很少有认知障碍的病情较轻的患者可能会有帮助。需要提高临床医生的认识,以便尽快做出明确诊断并尽早提供正确的治疗。