Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA, USA.
Department of Biology, Massachusetts Institute of Technology, Cambridge, MA, USA.
Nature. 2018 Dec;564(7734):141-145. doi: 10.1038/s41586-018-0758-y. Epub 2018 Nov 28.
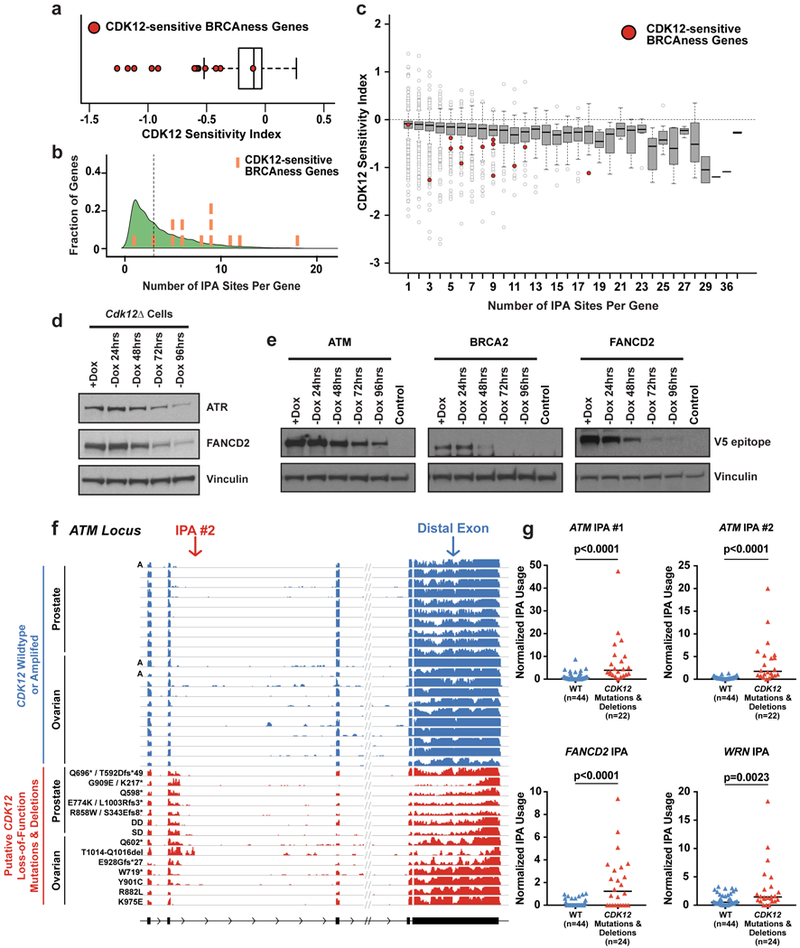
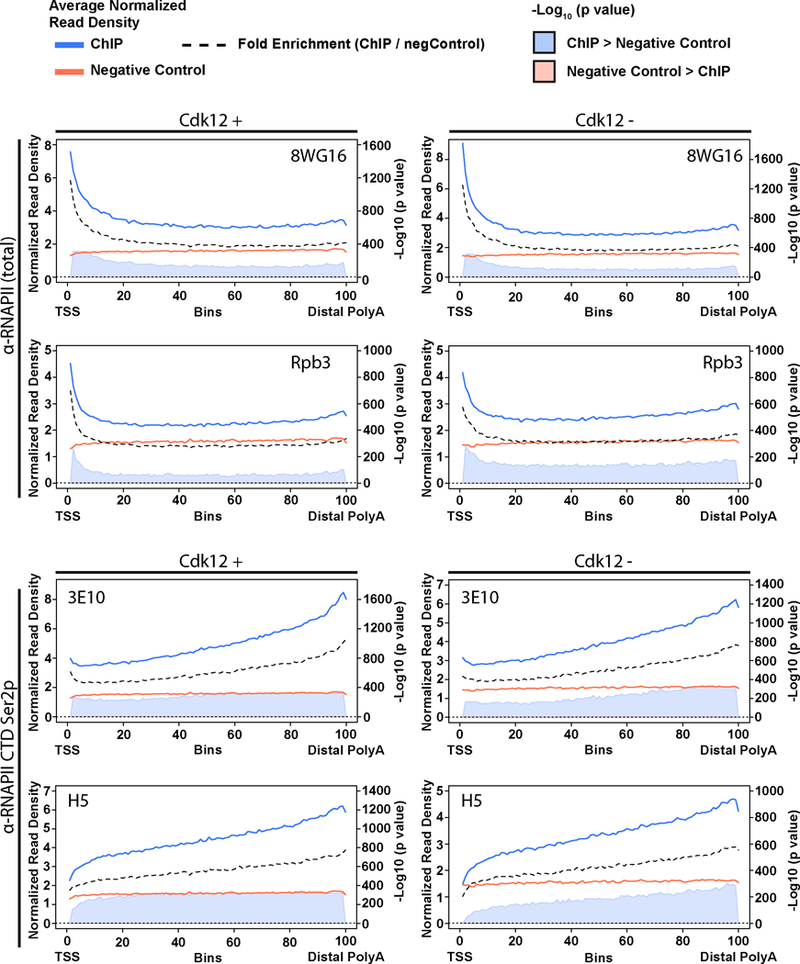
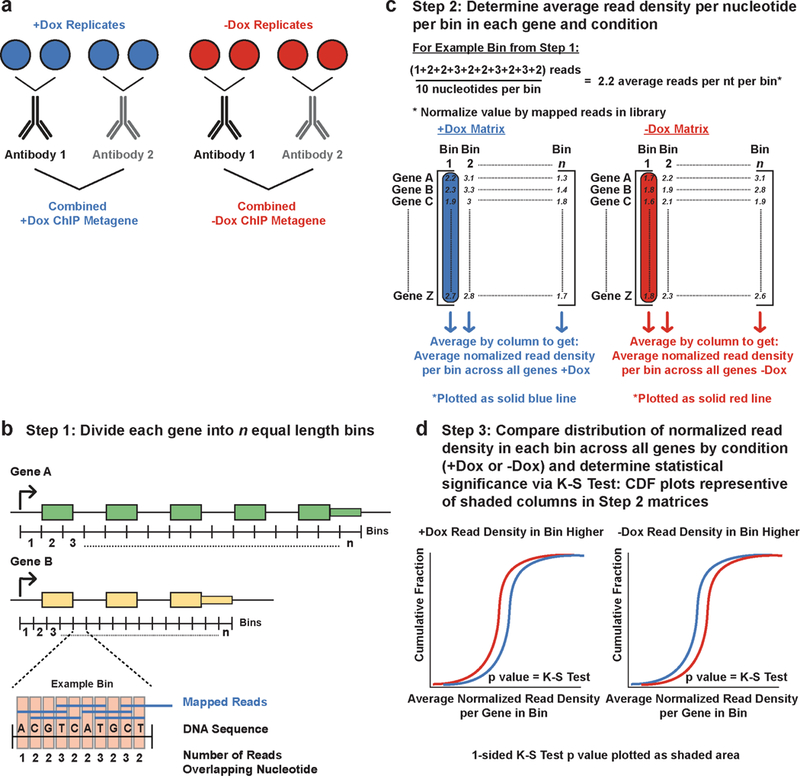
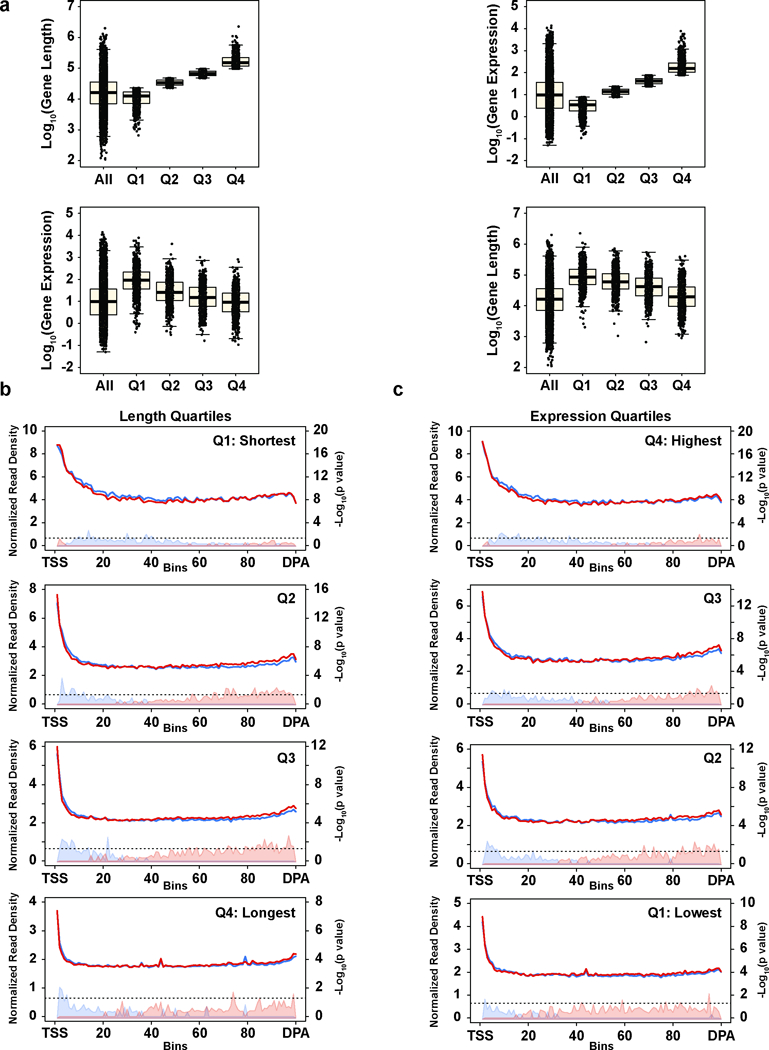
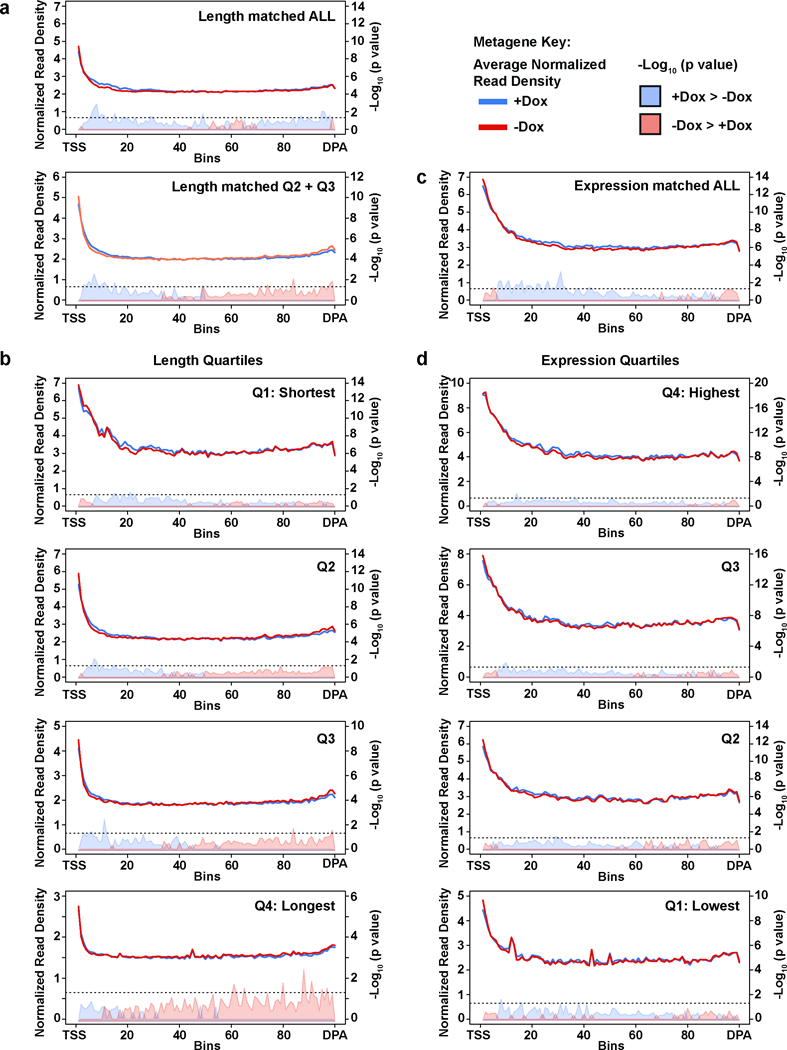
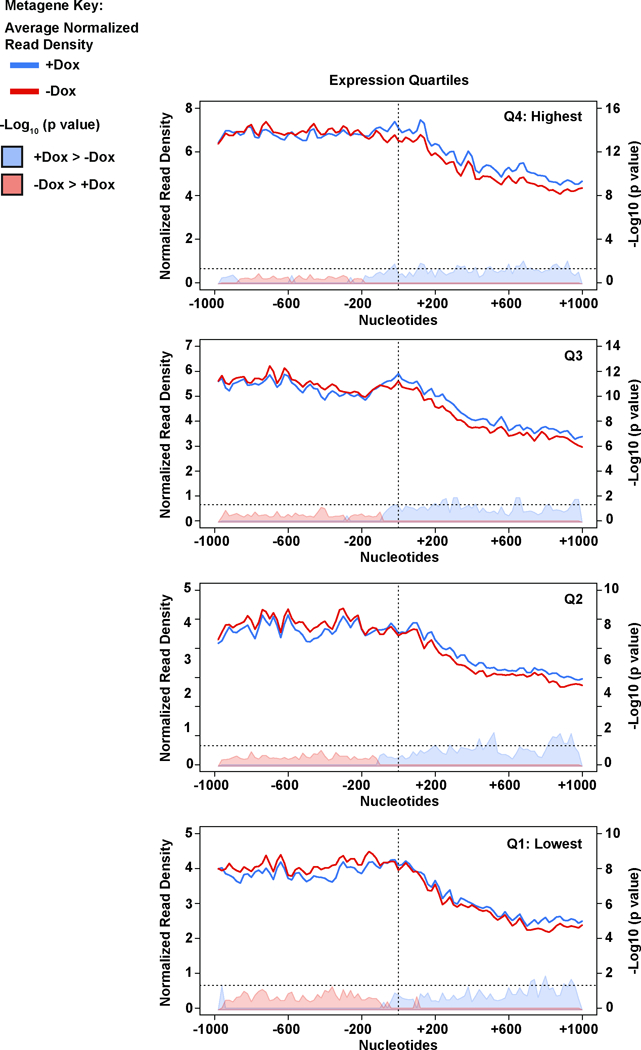
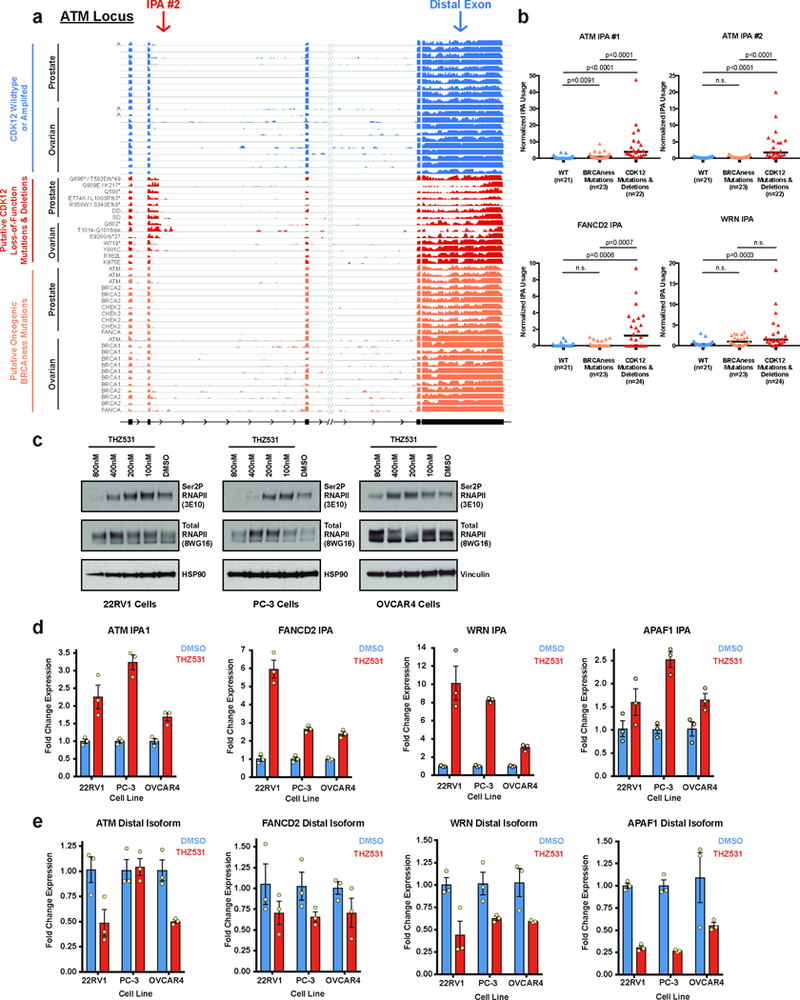
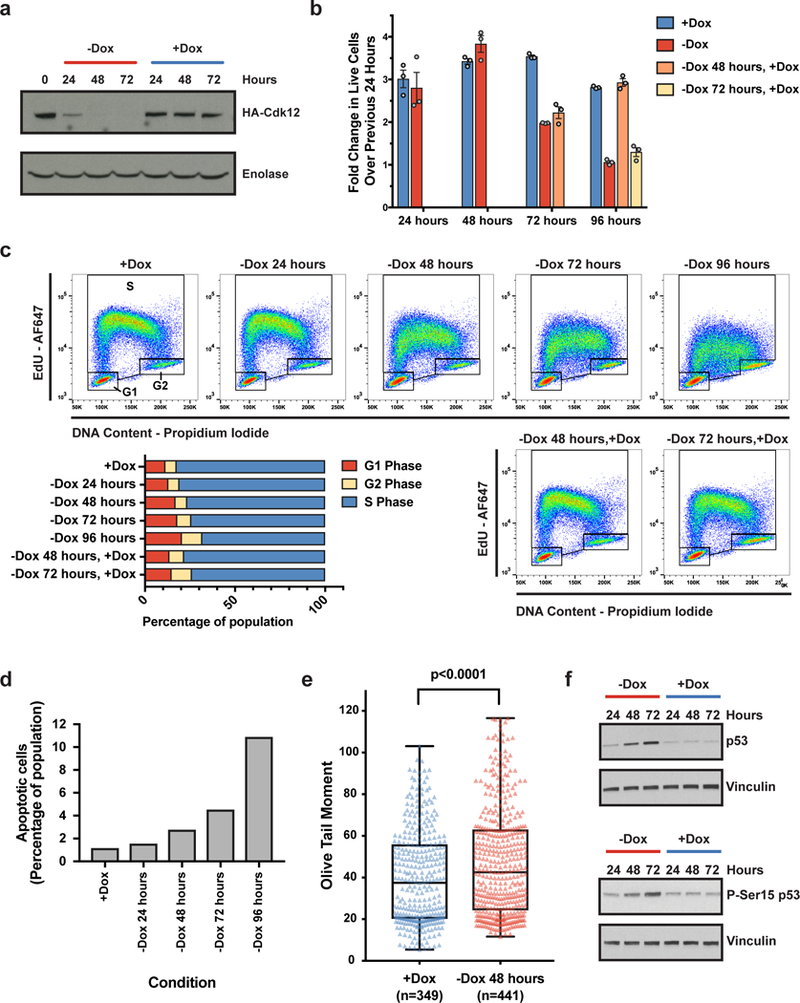
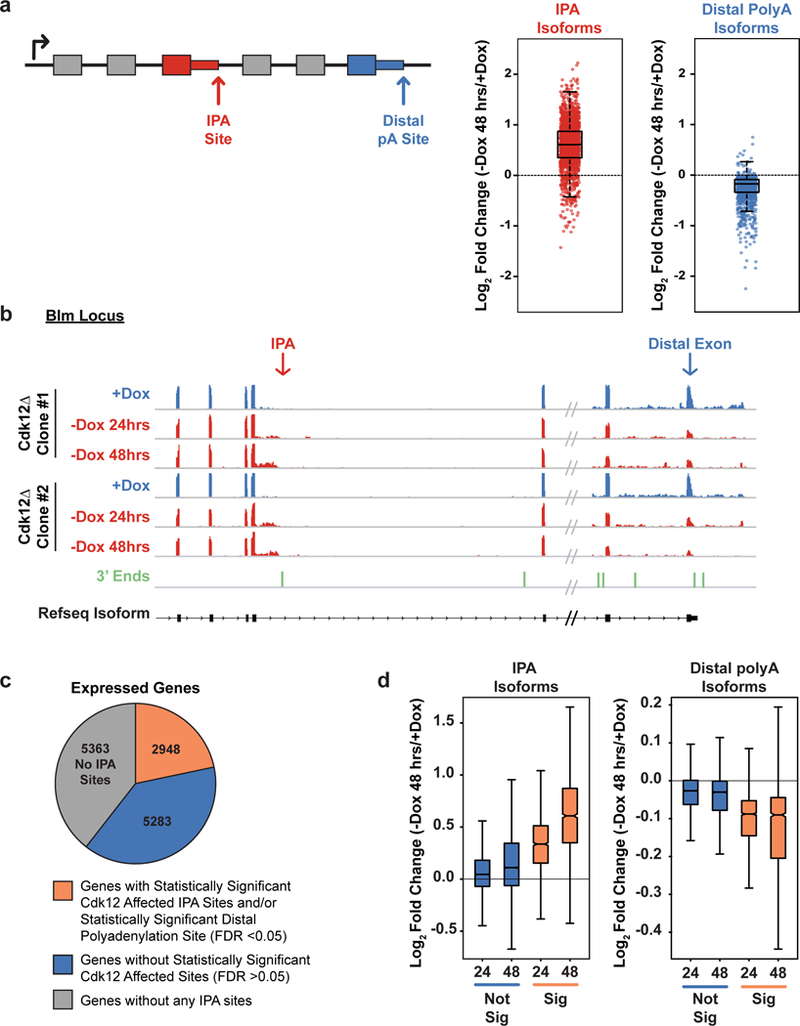
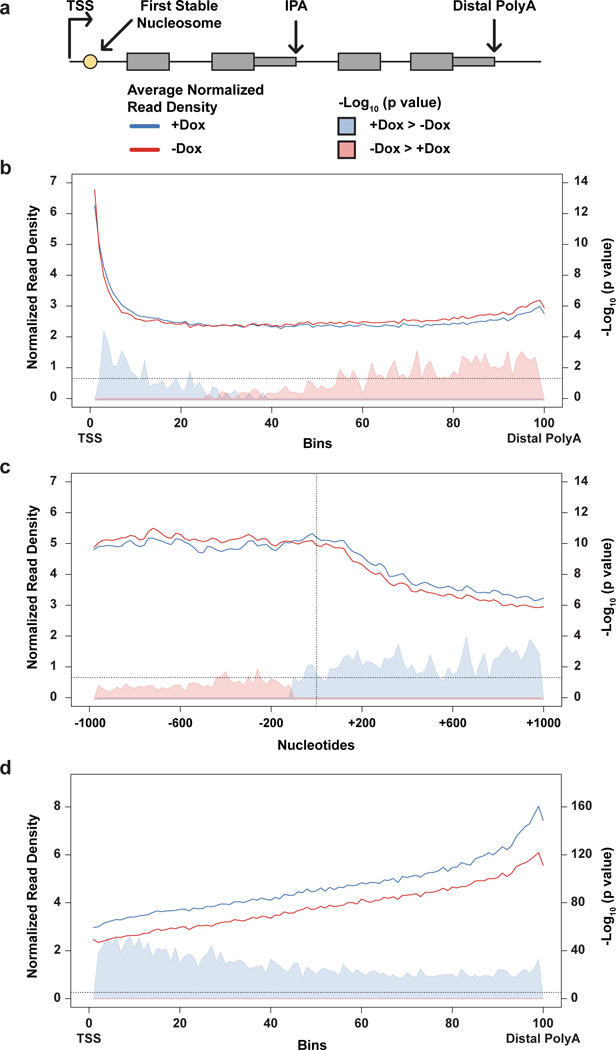
Mutations that attenuate homologous recombination (HR)-mediated repair promote tumorigenesis and sensitize cells to chemotherapeutics that cause replication fork collapse, a phenotype known as 'BRCAness'. BRCAness tumours arise from loss-of-function mutations in 22 genes. Of these genes, all but one (CDK12) function directly in the HR repair pathway. CDK12 phosphorylates serine 2 of the RNA polymerase II C-terminal domain heptapeptide repeat, a modification that regulates transcription elongation, splicing, and cleavage and polyadenylation. Genome-wide expression studies suggest that depletion of CDK12 abrogates the expression of several HR genes relatively specifically, thereby blunting HR repair. This observation suggests that the mutational status of CDK12 may predict sensitivity to targeted treatments against BRCAness, such as PARP1 inhibitors, and that CDK12 inhibitors may induce sensitization of HR-competent tumours to these treatments. Despite growing clinical interest, the mechanism by which CDK12 regulates HR genes remains unknown. Here we show that CDK12 globally suppresses intronic polyadenylation events in mouse embryonic stem cells, enabling the production of full-length gene products. Many HR genes harbour more intronic polyadenylation sites than other expressed genes, and these sites are particularly sensitive to loss of CDK12. The cumulative effect of these sites accounts for the enhanced sensitivity of HR gene expression to CDK12 loss, and we find that this mechanism is conserved in human tumours that contain loss-of-function CDK12 mutations. This work clarifies the function of CDK12 and underscores its potential both as a chemotherapeutic target and as a tumour biomarker.
削弱同源重组(HR)介导修复的突变促进肿瘤发生,并使细胞对导致复制叉崩溃的化疗药物敏感,这种表型称为“BRCAness”。BRCAness 肿瘤源自 22 个基因的功能丧失突变。在这些基因中,除了一个(CDK12)之外,所有基因都直接作用于 HR 修复途径。CDK12 磷酸化 RNA 聚合酶 II C 末端结构域七肽重复丝氨酸 2,这种修饰调节转录延伸、剪接和切割及多聚腺苷酸化。全基因组表达研究表明,CDK12 的耗竭相对特异地消除了几个 HR 基因的表达,从而削弱了 HR 修复。这一观察结果表明,CDK12 的突变状态可能预测对 BRCAness 的靶向治疗(如 PARP1 抑制剂)的敏感性,并且 CDK12 抑制剂可能诱导 HR 功能正常的肿瘤对这些治疗的敏感性。尽管临床兴趣日益增长,但 CDK12 调节 HR 基因的机制仍不清楚。在这里,我们表明 CDK12 全局抑制了小鼠胚胎干细胞中的内含子多聚腺苷酸化事件,从而产生全长基因产物。许多 HR 基因比其他表达基因具有更多的内含子多聚腺苷酸化位点,这些位点对 CDK12 的缺失特别敏感。这些位点的累积效应解释了 HR 基因表达对 CDK12 缺失的敏感性增强,我们发现该机制在含有功能丧失 CDK12 突变的人类肿瘤中是保守的。这项工作阐明了 CDK12 的功能,并强调了它作为化疗靶点和肿瘤生物标志物的潜在作用。