Department of Biomedical Engineering and Center for Biological Systems Engineering , Washington University in St. Louis , One Brookings Drive , Campus Box 1097, St. Louis , Missouri 63130 , United States.
J Chem Theory Comput. 2019 Feb 12;15(2):1355-1366. doi: 10.1021/acs.jctc.8b00572. Epub 2019 Jan 22.
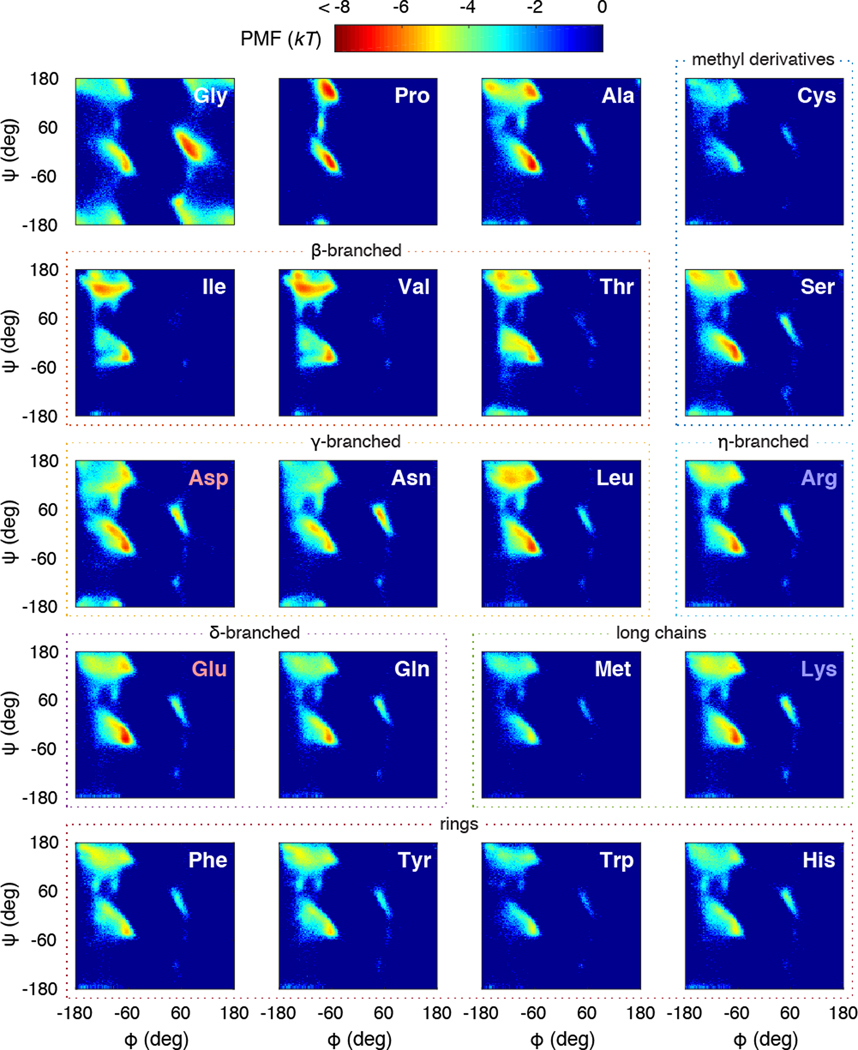
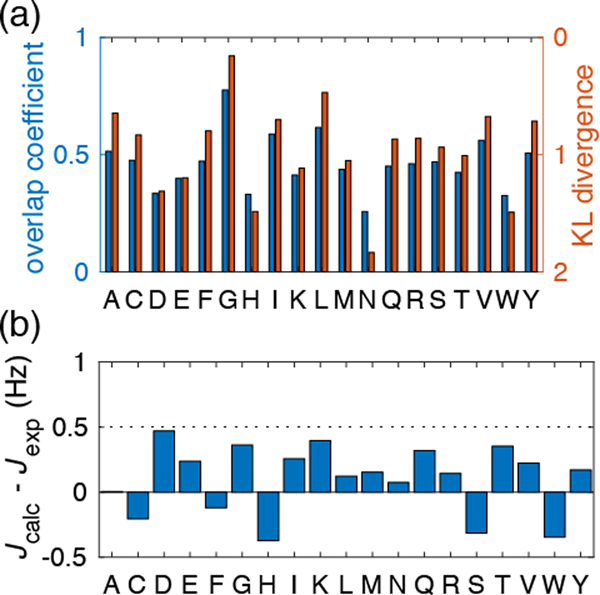
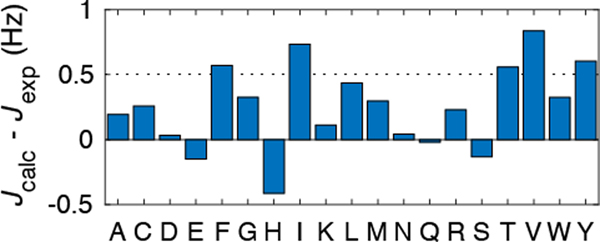
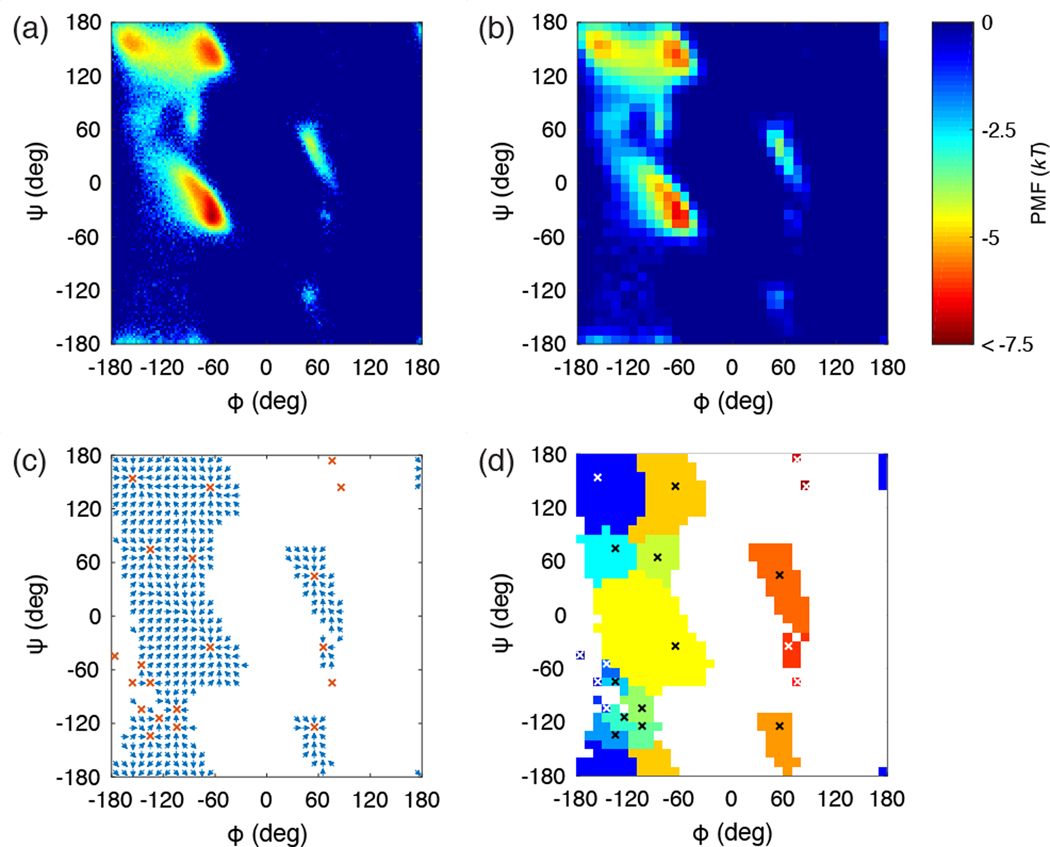
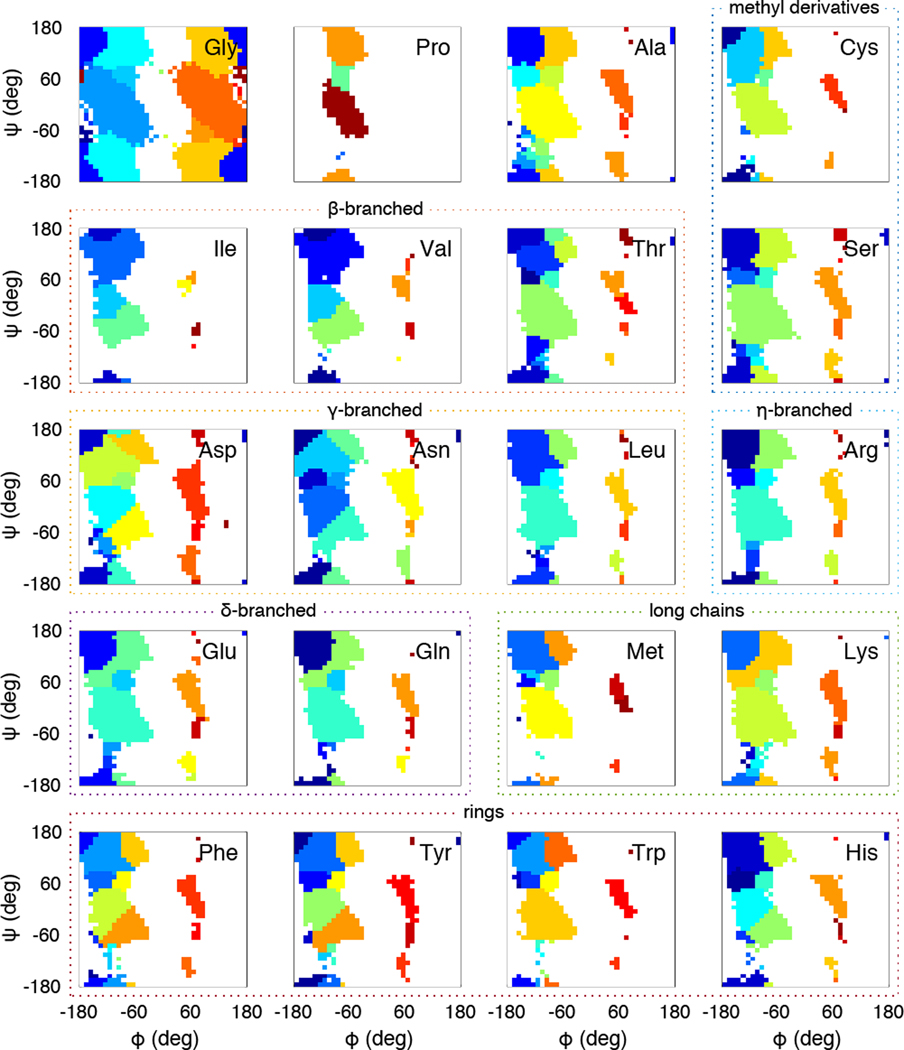
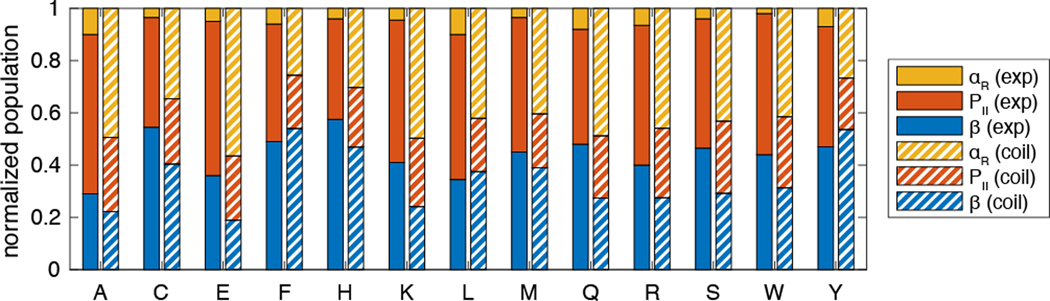
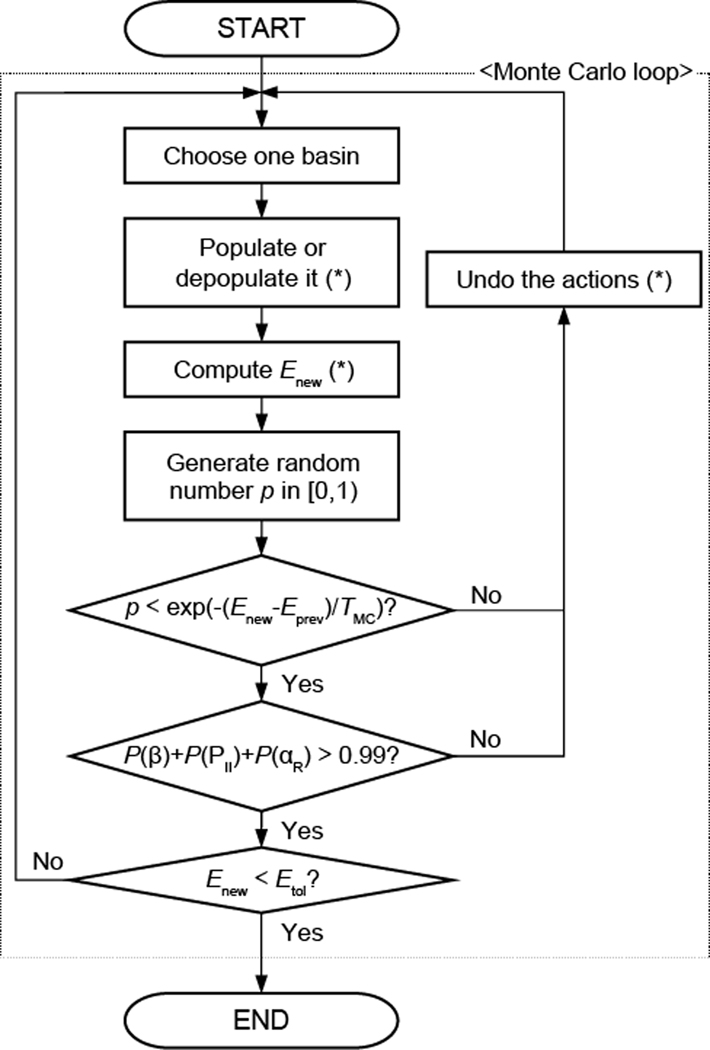
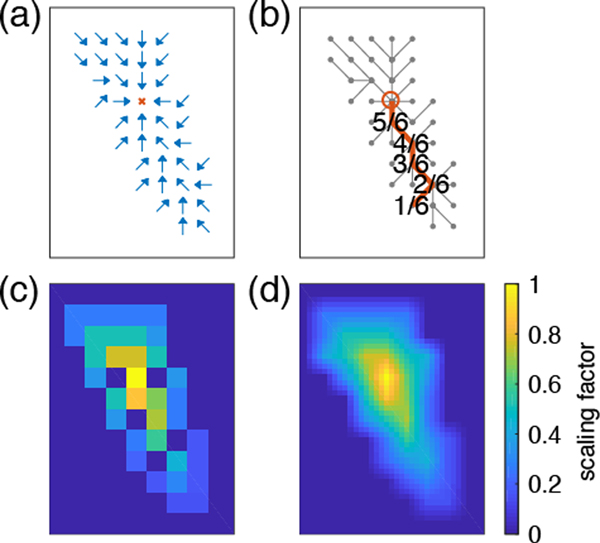
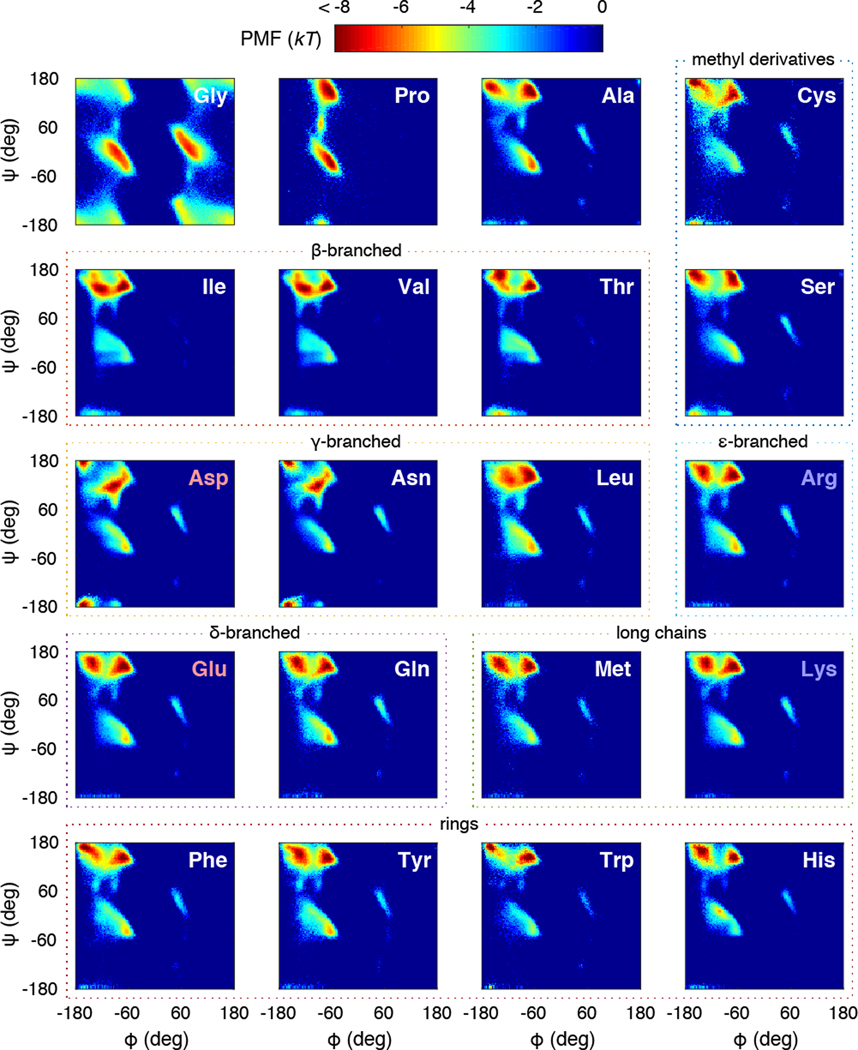
Experimentally derived, amino acid specific backbone dihedral angle distributions are invaluable for modeling data-driven conformational equilibria of proteins and for enabling quantitative assessments of the accuracies of molecular mechanics force fields. The protein coil library that is extracted from analysis of high-resolution structures of proteins has served as a useful proxy for quantifying intrinsic and context-dependent conformational distributions of amino acids. However, data that go into coil libraries will have hidden biases, and ad hoc procedures must be used to remove these biases. Here, we combine high-resolution biased information from protein structural databases with unbiased low-resolution information from spectroscopic measurements of blocked amino acids to obtain experimentally derived and computationally optimized coil-library landscapes for each of the 20 naturally occurring amino acids. Quantitative descriptions of conformational distributions require parsing of data into conformational basins with defined envelopes, centers, and statistical weights. We develop and deploy a numerical method to extract conformational basins. The weights of conformational basins are optimized to reproduce quantitative inferences drawn from spectroscopic experiments for blocked amino acids. The optimized distributions serve as touchstones for assessments of intrinsic conformational preferences and for quantitative comparisons of molecular mechanics force fields.
实验得出的、氨基酸特异性的主链二面角分布对于模拟蛋白质数据驱动的构象平衡以及评估分子力学力场的准确性非常有价值。从蛋白质高分辨率结构分析中提取的蛋白质卷曲库已被用作量化氨基酸固有和上下文相关构象分布的有用代理。然而,卷曲库中的数据会存在隐藏的偏差,必须使用特定的程序来消除这些偏差。在这里,我们将来自蛋白质结构数据库的高分辨率有偏信息与来自受阻氨基酸光谱测量的无偏低分辨率信息相结合,为 20 种天然存在的氨基酸中的每一种获得实验得出和计算优化的卷曲库图谱。构象分布的定量描述需要将数据解析为具有定义的包络、中心和统计权重的构象盆地。我们开发并部署了一种数值方法来提取构象盆地。构象盆地的权重经过优化,以再现对受阻氨基酸进行光谱实验得出的定量推论。优化后的分布可作为评估固有构象偏好和定量比较分子力学力场的基准。